A Complete Guide to Protein Folding Prediction with RoseTTAFold: Part I
A 3-hour breakdown of the three-track RoseTTAFold protein prediction model that leverages multi-modal deep learning architectures to transform single sequences into dynamic 3D structures.
(An interactive table of contents can be found on the left sidebar)
Introduction
Preface
The Structure of Proteins
Amino Acids
Protein Folding
Representing Protein Structure
Multiple Sequence Alignments
Hidden-Markov Model Profiles
Initializing the Emission Probabilities
Log-Odds Score with Viterbi Algorithm
Dynamic Programming Matrices
Log-Sum-Of-Odds Score
Column Score
Viterbi Algorithm for HMM-HMM Alignment
Secondary Structure Score
Correlation Score
SIMD and Vectorized Viterbi Algorithm
Iterative MSA Alignment
MSA Embeddings
1-Dimensional Positional Encoding
Query Encoding
Initializing the MSA Embeddings
Axial Attention
Multi-Head Axial Attention
Tied Axial Attention
Soft-Tied Residue-Wise Attention
Linearized Attention with Performer
Kernels and Bochner’s Theorem
Trigonometric Random Feature Estimator
Positive Random Feature Estimator
Orthogonal Random Features
Implementing the Softmax Random Feature Map
Multi-Head Fast Attention
Axial Encoder
Looking Foward
Introduction
If you’ve read my previous article on Spherical Equivariant Graph Transformers, you would know that my end goal was to fully understand the structure module of the RoseTTAFold protein prediction architecture introduced in the paper Accurate Prediction of Protein Structures and Interactions Using a Three-Track Neural Network. What differentiates RoseTTAFold from AlphaFold and earlier protein folding models is the bidirectional flow of features across distinct representations of proteins, from sequence-based MSA embeddings to pair-wise co-evolution maps to 3D geometric graphs.
If you want to extend your intuition behind attention-based deep learning architectures for multi-modal protein data, there is no better model to learn than RoseTTaFold (RF). Here, you will learn the theory and implementation of a diverse array of attention-based algorithms integrated into the RF architecture—including tied axial attention, fast attention with the Performer architecture, direct attention, graph transformers, SE(3)-Transformers, and masked attention—and ultimately see how they transform (pun intended) simple 1D sequences of letters into dynamic 3D structures that govern the fundamental processes of life.
Full three-track protein prediction architecture. The 1D track updates the MSA embeddings, the 2D track updates the pair features, and the 3D track updates a 3D geometric graph representation. (Source: Alchemy Bio)
Each track of the RF model captures 1D, 2D, and 3D feature-rich representations that continuously share features to enhance the accuracy of each representation. At a high level, the integration of features across tracks involves:
Generating Multiple Sequence Alignments (MSA) by comparing the Hidden Markov Model profiles of the target sequence with a database of template sequences. The sequences with the top scores are considered homologous alignments with similar structural and functional properties to the target and are added to the MSA.
Processing the MSA with residue-wise (row-wise) and sequence-wise (column-wise) axial attention to generate a contextual embedding for each residue in the MSA that captures dependencies across the sequence it belongs in and the aligned residues within each column of the MSA through tied attention weights.
Initializing the pair-wise feature map to capture interaction and co-evolution motifs for every pair of positions in the target sequence. An embedding for each pair is initialized with distance and orientation data from known homologous structures generated from the MSA search.
Updating the pair-wise feature map from the MSA embeddings by taking the weighted average of the embeddings across all N sequences for each position and computing the outer product between each pair of positions to get the co-evolution features.
Processing the pair-wise embeddings with axial attention along the rows and columns of the pair-wise feature map to capture dependencies across pair interactions with shared residues.
Updating the MSA embeddings with direct multi-head attention across the residue dimension using attention maps derived from transforming the pair-wise embeddings into attention scores.
Generating the initial coordinates of each residue for the geometric graph representation from a fully connected graph with MSA embeddings as node features and the pair features as edge features and applying the Graph Transformer.
Updating the geometric graph representation using SE(3)-Transformer blocks to update the 3D protein backbone coordinates and to generate a set of scalar state features for each residue.
Using the state features and the pair-wise distance between residues from the SE(3)-Transformer to update the MSA embeddings with masked direct multi-head attention on the MSA embeddings.
You may be wondering: why break down RoseTTAFold instead of AlphaFold or even the updated version, RoseTTAFold2?Here are some of the key reasons to convince you that RF is worth understanding.
A single iteration is all that’s needed. Although AlphaFold and even the new RoseTTAFold2 introduced the recycling mechanism that feeds output structural features as input to the next iteration, RF can generate comparably accurate predictions with just a single iteration through the model due to the parallelized, three-track approach.
The end goal is not just structure. Producing an accurate structural representation of a protein sequence can provide valuable insights into the function of unknown proteins, but the feature-rich MSA and pair-wise embeddings can also be useful for various downstream tasks such as binding affinity prediction, binder design, etc.
RF is capable of modeling protein-protein complexes and quaternary structures from sequence alone. Due to memory constraints, RF was trained to generate feature-rich MSA and pair-wise embeddings of several discontinuous protein subunits and combine them into a full protein structure using a final SE(3)-Transformer block. Thus, RF is capable of generating the backbone coordinates of the protein-protein complexes from multiple input sequences, bypassing the need for intermediate structure prediction of each sequence independently and predicting binding on the rigid subunit structures.
RF can convert raw sequences to 3D coordinates with limited template structures. The updated RF2 removed the two-track block and replaced it with a single three-track block that immediately integrates the template structure at the beginning of the model. Although the original RF model initializes the pair-wise embeddings with template structure information, the two-track block generates feature-rich embeddings without close structural homologs by relying heavily on the MSA embeddings that are then converted into initial 3D coordinates through the Graph Transformer network (removed in RF2).
In this post, I will introduce all the fundamental concepts of protein-folding prediction models including multiple-sequence alignments, multi-modal feature processing, and loss functions, and how all these concepts come together in the RF architecture. This will be a long journey, so let’s dive in.
Preface
Summaries and additional useful information (e.g. proofs, background theory, etc.) that is not nessesary in the understanding of the model will be placed in these quotation blocks.
Here, I have listed the notation used throughout the article to denote different variables and dimensions that closely align with the notation used throughout the original paper and related literature:
q → the query or target sequence (the sequence that we are predicting the structure for).
t → template sequence of homologous proteins in the MSA.
B → number of query sequences in a training batch for stochastic gradient descent.
N → number of template sequences in the MSA.
L → number of columns in the MSA alignment which represent positions in the amino acid sequence.
i, j → amino acid positions along the residue dimension of the MSA indexed from 1 to L. Position i often refers to the position that attention is computed for and position j is the position that position i is attending to.
n, m → indices of the template sequence along the sequence dimension of the MSA indexed from 1 to N.
H → number of attention heads for multi-head attention.
q, k, v → query, key, and value embeddings for computing attention (bolded to indicate that they are vectors and to differentiate from q).
Q, K, V → matrices of stacked query, key, and vector embeddings stacked along the rows of the matrix.
d_msa → dimension of the MSA embeddings.
d_pair → dimension of the pair-wise embeddings.
d_temp → dimension of the template embeddings.
d_h → dimension of embeddings split for multi-head attention.
d_model → dimension of the embeddings for general classes. Since several modules are used across different representations, this could take the values of d_msa, d_pair, d_temp, etc.
r_ff → multiplier for the feed-forward layers. If r_ff = 4, then the dimension of embedding d is projected to an intermediate dimension of 4*d before being projected back down to d.
exp(x) → exponential e^x
||x|| → L2 norm or Euclidean distance of a vector x computed by taking the square root of the sum of all squared elements along the vector dimension.
\(||\mathbf{x}||=\sqrt{x_1^2+x_2^2+\dots x_d^2}\)
Many more pieces of notation will be introduced at specific sections of the articlebut this is the general set of notation that will be used throughout.
Furthermore, to avoid redundancy in the code explanations, here is a breakdown of some functions (and their PyTorch implementations) that will be used extensively throughout the architecture. Note that some of these are built-in PyTorch modules or functions, while others are implemented specifically for RF.
Dropout layer (nn.Dropout(p_drop)) → built-in PyTorch module that applies dropout regularization, randomly sets entries along the embedding dimensions to 0 with a probability of p_drop (usually 0.1) to avoid overfitting to training examples.
Linear transformation layer (nn.Linear(d_in, d_out)) → built-in Pytorch module that transforms an array with shape (…, L, d_in) to an array with shape (…, L, d_out) by multiplying each L x d_in matrix stored in the last two dimensions by a distinct d_in x d_out matrix of learnable weights and adding a d_out-dimensional vector of learnable weights (or bias vector). This outputs an array with shape (…, L, d_out) where the last two dimensions are L x d_out matrices of transformed feature embeddings.
Normalization layer (LayerNorm(d_model))→ module that normalizes each feature embedding x to have an approximate mean of 0 by subtracting the mean of the entries and a variance of 1 by dividing by the standard deviation. This implementation also introduces a learnable multiplier, a, and a learnable bias vector, b.
Feed-forward layer (FeedFowardLayer(d_model, d_ff)) → module that applies a sequence of two linear transformation layers: the first projects each embedding to a higher dimensional feature space with dimension d_ff, applies dropout and a ReLU nonlinear activation function, and the second projects each embedding back down to the original feature space with dimension d_model. This is a simple feature-processing layer used to capture nonlinear relationships between features in a higher-dimensional space.
Einsum function (torch.einsum) → built-in PyTorch function that simplifies computation of various matrix and vector operations using Einstein notation. This function takes as input an equation string that specifies the dimensions of the input arrays and the dimensions of the output array as well as the specific operands in the order that they appear in the equation string. The function computes the element-wise product of the operands aligned based on the input dimensions specified in the equation and sums over the dimensions not included in the output dimensions. The “…” symbol indicates dimensions that are already aligned in the input. To build a foundation for some functions that will be used later, I have given the implementations of some common matrix and vector operations using torch.einsum. You can also refer to the documentation here.
In addition, here is a compilation of tensor operations in PyTorch that are used to manipulate the size and order of dimensions in a PyTorch tensor.
In this article, I’ll refer to PyTorch tensors as ‘arrays’ to remain consistent with my previous article on SE(3)-Transformers that makes use of geometric tensors (spherical tensors) and to avoid confusion in Part 2 when we discuss the application of SE(3)-Transformers in the RF architecture.
The dimensions of an array are referred to as its ‘shape’ which will be given in parentheses as a list of integers indicating the size of each dimension. For instance, an array of shape (L, d) holds a total of L*d elements.
Throughout the article, I have included code from the full PyTorch implementation of the RoseTTAFold model on GitHub that I fully annotated and made slight modifications for clarity.
I’ll be breaking down most of the classes but placing more emphasis on the mathematics and intuition surrounding the code, so I encourage you to refer to the full implementation to connect the snippets and intuition you gain from this article to the full functions and classes.
The Structure of Proteins
Proteins are sequences of multi-atomic molecules called amino acid residues from a total of 20 amino acid types composed of a backbone that is constant across types and a side chain that is unique to each type of amino acid. The primary protein sequence is formed by covalent peptide bonds along the amino acid backbone, but the complex 3D structure of proteins is dependent on covalent and non-covalent interactions between side chains from distant residues in the sequence.
Amino Acids
Proteins are sequences of amino acid residues. There are 20 standard types of amino acids, each consisting of the same set of backbone atoms and a unique set of atoms called the R-group (or side chain).
The general structure of a single amino acid. For each amino acid type, there is a unique set of atoms in the location of the R-group. (Source: Alchemy Bio)
The backbone atoms include an amino group (-NH2) also called the N-terminus and a carboxyl group (-COOH) called the C-terminus. These groups are connected to either side of a central carbon atom called the C-alpha (Cα) atom.
The R-group or side chain connected to the Cα atom is unique for each type of amino acid. The identity of the R-group gives each type of amino acid its distinct properties including hydrophobicity/hydrophilicity, charge, pH, and structure. These properties determine the covalent and non-covalent bonding interactions that occur between amino acids in the sequence which cause the sequence to fold into the most thermodynamically favorable conformation.
All amino acids except for Glyceine have a carbon atom connecting the Cα atom and the R-group, which is called the Cβ atom. The positioning of the Cβ atoms of residues across the sequence is useful in describing the orientation and distance of residues relative to each other in the folded protein structure.
The 20 standard and 2 special amino acids are categorized by their properties. The unique R-groups are highlighted in color. (Source)
Protein Folding
Protein folding involves multiple stages: primary structure, secondary structure, tertiary structure, and sometimes quarternary structure.
Stages of protein folding from primary structure (amino acid sequence), secondary structure (alpha-helix, beta-sheets, etc.), tertiary structure (3D conformation), and quaternary structure (multi-polypeptide complexes). (Source)
The primary structure is the sequence of amino acids of the polypeptide chain that is formed by covalent peptide bonds between the carboxyl group (-COOH) at the C-terminus and the amino group (-NH2) at the N-terminus of adjacent amino acids in the sequence. The sequence is the primary input to structure prediction models.
Formation of a peptide bond between two amino acids. Primary protein structures are sequences of amino acids connected by peptide bonds. (Source: Alchemy Bio)
The secondary structure is formed directly from the sequence through hydrogen bonding between the atoms in the polypeptide backbone from the electrostatic attractions between the partially negative oxygen atom at the C-terminus of one residue and the partially positive hydrogen atom at the N-terminus of another residue. The most common secondary structure states include α-helixes and β-sheets.
Representation of α-helixes and β-sheets in protein structure models (Source)
The tertiary structure is formed from interactions between R-groups or side chains of the residues in the sequence. Since the side chains for each type of amino acid have unique chemical properties (charge, hydrophobicity, polarity, etc.), distinct sequences of residues form unique 3D structures that characterize a protein. Interactions include non-covalent bonds like hydrogen bonds, ionic bonds, dipole-dipole bonds, hydrophobic interactions, van der Waal forces, and a special type of covalent disulfide bond that forms between two cysteine residues.
Formation of tertiary structure from non-covalent (hydrogen bonds, ionic bonds, hydrophobic interactions) and covalent (disulfide linkage between cysteine residues) interactions between amino acid side-chains (Source).
The quaternary structure is only present in some proteins that are formed from multiple disconnected polypeptide chains that form subunits that come together after folding to form a higher-level structure. When tasked to predict quaternary structures of multi-sequence complexes, RF performed exceptionally well with template modeling (TM)-scores ~90, indicating high similarity to the true structures.
Quarternary structures predicted from the RoseTTAFold model of proteins with two (A) and three (B) polypeptide chains. In the two-subunit proteins shown in (A), the first subunit is colored grey and the second is multicolored (blue is the N-terminus, and red is the C-terminus). In the three-subunit structures shown in (B), the separate subunits are colored grey, magenta, and cyan. The experimentally determined structures (left) and the model predictions (right) have template modeling scores >80, indicating high structural similarity (Source).
Representing Protein Structure
RF defines a set of distances and anglesthat fully define the spatial relationships between each pair of residues and their constituent atoms. The model is trained on data containing ground-truth labels for each of these structural features to accuractly predict the features for unknown protein sequences. This representation is defined such that we can unambiguously derive the full protein structure.
RF defines a set of four features defining the relative spatial coordinates for every ordered pair of residues in the protein sequence, including measures of distance, bond angles, and dihedral angles.
Set of four structural features defining the relative spatial position of every ordered pair of residues (i, j), including the distance between sidechains, the dihedral angles defining the relative orientation of atoms, and the bond angle. (Source: Alchemy Bio)
Before we describe each of the four features, let’s first clarify the definition of a dihedral angle.
A dihedral angle defined by four points A-B-C-D is the angle from the plane formed by coordinates A-B-C to the plane formed by coodinates B-C-D around the vector connecting B to C.
As shown in the diagram below, the dihedral angle is equivalent to the angle between the normal vector m to the plane spanned by A-B-C and the normal vector n to the plane spanned by B-C-D.
A diagram of the dihedral angle between points A-B-C-D about the line connecting B-C. This is the angle from the plane containing points A-B-C and the plane containing points B-C-D and can be computed with the normal vectors to the planes m and n. (Source)
The dihedral angle takes values from [-π, π], where the sign indicates the direction of rotation around the central vector pointing from B to C.
We can visualize the sign of the dihedral angle as the direction of rotation of the second plane defined by points B-C-D from the first plane defined by points A-B-C if they start aligned such that their normal vectors are parallel. The dihedral angle is positive when the second plane is rotated by an angle of 0 and π in the direction of the normal vector of the first plane and negative when it is rotated in the direction opposite to the normal vector. In the image above, the dihedral angle is positive since the plane B-C-D is rotated in the same direction as the normal vector m.
When describing protein structures, the sign of the dihedral angles are crucial since it determines the direction of rotation around the central bond relative to the other atoms in the protein not including the four atoms defining the planes.
The set of four features for an ordered pair of residues i and j in the protein sequence (which we will refer to as the position of i with respect to j) include:
The scalardistance d_ij between the Cβ atom of residue i and the Cβ atom of residue j or the length of the virtual bond connecting the Cβ~Cβ atoms. This measures the distance between the side chains of the two residues, where the non-covalent interactions between residues occur.
We can compute d by subtracting the vector to the Cβ atom of residue j from the vector to the Cβ atom of residue j and computing the length of the displacement vector (L2 norm).
\(d_{ij}=d_{ji}=\underbrace{||\mathbf{x}_i^{C\beta}-\mathbf{x}_j^{C\beta}||}_{\text{length of displacement vector between C}\alpha \text{ atoms}}\)
Scalar distance d between the Cβ atoms from both residues in the pair. This measures the distance between the residue sidechains, which is the typical site of non-covalent interaction between residues across the protein. (Source: Alchemy Bio)
The dihedral angle ω_ij from the plane formed by the i(Cα)-i(Cβ)~j(Cβ) atoms to the plane formed by the i(Cβ)~j(Cβ)-j(Cα) atoms, where ~ indicates the imaginary bond between the Cβ atoms of the two residues. This angle determines the orientation of the central Cα atoms relative to each other.
To compute the dihedral angle, we first compute the three displacement vectors between the four atoms pointing in the direction from i(Cα) to j(Cα).
Then, we use the displacement vectors to compute the unit normal vectors perpendicular to each of the planes by taking the cross product of the vectors that span the plane. Since we are computing the dihedral angle for residue i with respect to j, we define the the plane with normal vector n_1 to be the plane spanning i(Cα)-i(Cβ)~j(Cβ) which will determine the sign of the dihedral angle.
Dihedral angles can range from [-π, π] defined by the direction of rotation around the central bond (Cβ~Cβ for ω_ij) from the first plane spanning residue i to the second plane spanning residue j. To determine the sign, we compute the polar sine from the dot product of the vector of the central bond d_ij onto the cross product between n_1 and n_2, which is also the signed area of the parallelopiped formed by the three vectors.
\( \sin(\omega_{ij})=\underbrace{\frac{\mathbf{d}_{ij}\cdot(\mathbf{n}_1\times \mathbf{n}_2)}{||\mathbf{d}_{ij}||||\mathbf{n}_1||||\mathbf{n}_2||}}_{\text{polar sine component of }\omega_{ij}}\)
This angle value is positive when the direction of rotation of the second plane n_2 around the central bondis in the direction of n_1, and negative otherwise. To convert this into an angle in the range [-π, π], we compute the arctangent with respect to cosine component and the polar sine component.
This method computes the unique dihedral angle with the correct sign and range.
ω_ij dihedral angle around the Cβ~Cβ virtual bond connecting the sidechain carbon atoms of residue i and j. (Source: Alchemy Bio)
The dihedral angle θ_ij from the plane of the i(N)-i(Cα)-i(Cβ) atoms in residue i to the plane of the i(Cα)-i(Cβ)~j(Cβ) atoms bonded to residue j. This angle determines the relative orientation of the terminal nitrogen atom of residue i with respect to the side chain of residue j.
Thi sangle is computed with the same process as ω_ij, except with different vectors defining the planes.
θ_ij dihedral angle around the Cα-Cβ bond that determines the relative orientation of the terminal N in residue i from the side chain of residue j. (Source: Alchemy Bio)
The planar bond angle φ_ij from the Cβ-Cα bond in residue i to the virtual bond connecting the Cβ~Cβ atoms between i and j, which defines the position of the Cβ atom of the residue j relative to the reference frame centered at residue i.
To compute the planar bond angle, we use the equation for the dot product between the two displacement vectors from the central atom that form the angle:
The planar bond angle from the Cβ-Cα bond in residue i to the virtual bond connecting the Cβ~Cβ atoms between i and j (Source: Alchemy Bio)
These features may differ depending on the residue we are centered at. If we were instead centered at residue j, even though the distance between side chains is the same, the sign of the dihedral angle ω_ji will be flipped and the values of the angles θ_ji and φ_ij will be completely different.
Therefore, RF computes the set of features for every ordered pair of residues in the query sequence and stacks them into a pair-wise distance and orientation map with shape (B, L, L, 4).
Later, we will see how these features are incorporated into template embeddings and loss functions.
Multiple Sequence Alignments
Multiple Sequence Alignments (MSA) are matrices where the input query sequence is aligned with a stack of homologous template sequences to enable sequence-based predictions of protein properties (e.g. teritary structure, functional sites, or interaction sites) that are conserved across evolution.
Protein function directly correlates with its static or dynamic 3-dimensional structure, so proteins with the same function across two distant species can have different sequences due to evolutionary mutations but retain their tertiary structure and properties like functional sites and interaction interfaces. These proteins are called homologs.
Multiple Sequence Alignment (MSA) of the interleukin-12 receptor–interleukin-12 complex. Conserved regions are highlighted with color and deletions/insertions are denoted with ‘-’. (Source: Fig. S11 of Supplementary Materials)
Multiple Sequence Alignment (MSA) leverages the structural similarities across homologs by aligning homologous sequences with the query sequence to capture contextual patterns across homologs which can be used to predict structure from sequences alone and to generate features for an unknown query sequence from known homologous structures.
The MSA is an N (number of sequences) x L (length of the target sequence) matrix, where the first row is the query protein sequence and all the template sequences are stacked below. By comparing the residues along a column of the MSA, the model can identify conserved, co-evolved, and variable positions.
At a conserved position, the amino acid residue is the same across most homologs, indicating that the residue likely contributes to protein folding and function.
Co-evolved positions are pairs of columns that show correlated mutations with similar residues, indicating that the two positions in the query sequence likely interact during protein folding. If two positions have a strong interaction that contributes to protein function, a mutation in one position should be followed by a mutation in the other position such that their interaction properties remain constant.
At a variable position, the amino acid residues vary significantly across homologs, indicating that the position is irrelevant to protein structure and function.
MSAs contain conserved positions where the residues are the same across aligned homologs, coevolved positions where pairs of residues along the sequence depend on each other, and variable positions where the amino acids vary greatly across alignments. ‘-’ symbols represent gaps in the MSA. (Source)
Since mutations can occur not only as changes in residues but also as insertions or deletions (indels) between aligned sequences, the MSA includes indel gaps (represented as ‘-’ in the sequence) to align conserved and co-evolved positions that are offset across homologs.
MSAs are used to generate sequence profiles that describe the probabilities of all 20 types of amino acids appearing at each position in the sequence. These profiles are used in an iterative loop to search large databases for template sequences through sequence-to-profile or profile-to-profile alignment algorithms.
First, we will learn the fundamental theory behind HHsearch, an algorithm using Hidden-Markov Model (HMM) profiles for profile-to-profile alignment, before we discuss the computational speed-ups introduced in HHblits, and finally how these two algorithms work together to generate the final MSA for the query sequence.
Hidden-Markov Model Profiles
Hidden Markov Models (HMMs) are powerful probabalistic models that are used as sequence profiles to increase the sensitivity of alignment algorithms. A HMM sequence profile encodes information about the family of proteins that the sequence belongs in, including the probability that each type of amino acid will appear at a given position across the protein family and position-wise transition probabilities.
There are several databases with millions of protein sequences that cover the entire protein space, however, the databases of known protein structures are much more sparse, making it difficult to find homologs with relevant template structures by directly comparing the raw query and template sequences.
It turns out that even distant homologs with low sequence alignment with the query often share conserved structural motifs and are useful structural references for prediction. To identify these distant homologs, we need to construct sequence profiles that encode not only the raw sequence but also the position-wise mutation probabilities across its protein family.
At a high level, HMMs are probabilistic models that consist of emission probabilities for a set of states and transition (or conditional) probabilities between states. Given a sequence of observations, the HMM generates a probability score that determines the likelihood of those observations to occur based on the data used to construct the model.
The HMM profile of a query sequence is generated from an initial query MSA where each column of the MSA is converted into a column in the query HMM. Given a template sequence, the query HMM generates a probability score that determines how likely the template sequence is homologous to the query sequence.
The sensitivity introduced with HMM profiles enable HHsearch and HHblits to detect homologs where less than 20% of the residues exactly match the query sequence (<20% sequence identity). This demonstrates the insensitivity of sequence identity as a measure of homology. Instead, the estimated probability score generated by sequence-to-HMM and HMM-to-HMM alignments can detect homology with greater accuracy and sensitivity.
Hidden Markov Model (HMM) profile for a template sequence generated by an alignment of sequence homologs. Each match state (purple) has a unique probability distribution generated from the initial MSA. At each match state, we can transition into an insertion (pink), deletion (white), or the next match state. Allowed transitions between states are shown by the arrows. (Source: Alchemy Bio)
The query HMM profile contains three types of possible states at each position in the alignment: match (M), insertion (I), and deletion (D) states. Each state represents a type of alignment between the query HMM and a template sequence.
The match state (M) represents a position where the template sequence is aligned with the query sequence. Each residue aligned with a match state is assigned an emission probability q_i(x_l) indicating whether the residue is likely to be homologous to the query.
The emission probabilities for each type of amino acid at a match state is computed by counting the frequency of that type along the corresponding column of the MSA and dividing by the total number of sequences (rows) in the MSA. This generates a 20-dimensional probability distribution for each position in the HMM (represented by the size of the letters in the figure above). Smooth probability distributions indicate that a position is variable and do not require alignment whereas sharp distributions with probabilities close to 1 indicate that a position is conserved and should be aligned.
The insertion state (I) represents a position where the template sequence has extra residues between match states that don’t have a homologous partner in the template sequence. Each residue aligned with an insertion state is assigned the emission probability from a fixed background frequency f(a).
The emission probabilities of each amino acid type is equal to their background frequency f(a) equal to its frequency in all natural proteins. Common amino acids have high f(a) probabiltiies and rare amino acids have low f(a) probabilities.
The deletion state (D) represents a position where the template sequence has missing residues between match states and the query sequence has extra residues without a homologous partner in the template sequence. Since deletion states are not associated with an amino acid, there is no emission probability.
In addition to emission probabilities, template HMMs have transition probabilities defined for all allowed transitions between states through the HMM. The transition probabilities operate under the Markov assumption, which states that only the current state or position contributes to the probability of moving to the next state.
Match states can transition to another match state, an insertion, or a deletion state. The transition probabilities are distinct for each column of the HMM and depend on how frequently insertions and deletions occur in the next position in the MSA alignment.
Insertion states can transition back to the next match state or another insertion state. Each insertion state results in an increase in the length of the alignment by 1.
Deletion states can transition to a match state after skipping a match state or transition to another deletion state. Each deletion state results in a skipped match state.
The inclusion of transition probabilities in HMM profiles increases the sensitivity of alignment algorithms by acting as position-wise gap penalties that reduce the penalty for alignment gaps at locations with high insertion or deletion probabilities and increase penalties for alignment gaps at highly conserved positions, while other alignment algorithms assign a fixed gap penalty across all positions. This follows from the idea that insertions and deletions tend to occur at the same positions across homologs.
Given that HMM profiles can be used for sensitive MSA alignments, how do we construct the initial HMM?
Initializing the Emission Probabilities
To construct the initial HMM profile from a query or template sequence and update the emission probabilities given an expanded MSA, we define a procedure that leverages position-wise substitution matrices and sequence weights to compute the amino acid emission distribution at each column of the HMM.
From a single sequence, we can initialize an HMM profile by leveraging a position-wise substitution matrix.
A position-wise substitution matrix is a 20 x 20 matrix of scores (or pseudo counts) for each pair of the 20 total amino acid types, where each entry (a, x) is the likelihood of an amino acid of type a being replaced with a residue of type x based on known evolutionary mutation patterns.
The higher the likelihood of a type a amino acid being mutated into a type x amino acid, the higher the score in the substitution matrix.
HHsearch uses the Gonnetsubstitution matrix, a 20 x 20 lower-triangular matrix with positive entries indicating a high likelihood of replacement and negative entries indicating a low likelihood of replacement between the two residue types. Since it is a lower-triangular matrix, the score for type a being replaced with type x equals the score for type x being replaced with type a.
Gonnet substitution matrix. Higher scores indicate that the pair of amino acid types are more likely to be replaced by each other during evolution. (Source)
Diagonal entries (a, a) of the Gonnet matrix are always positive and are relatively higher if a has a high likelihood of remaining unchanged across evolution. For instance, Cysteine is the only amino acid type that forms disulfide bonds, so it is unlikely to mutate giving it a high self-replacement score of 11.5.
To turn the substitution scores into probabilities that can be used as initial emission probabilities, we first convert the Gonent matrix into a symmetric matrix by adding its transpose (excluding the diagonal entries) and applying the softmax function across each column to generate a set of 20 replacement probabilities for each of the 20 amino acid types.
An entry (a, x) of the resulting matrix is the probability of residue a being replaced with residue x and vice versa, normalized such that the set of 20 replacement probabilities for a single type x being replaced each of the 20 residue types sum to 1.
Therefore, if position i of the query sequence is a residue of type x, we can initialize the emission probabilities for column i of the query HMM using column x of the substitution probability matrix.
Even though these initial probabilities carry no contextual information about the given sequence, it is used to construct a baseline HMM profile from which we can iteratively refine as homologs are added to the MSA.
To do this, HHsearch updates the emission probabilities with the amino acid distribution across homologs unique to the query sequence as more template sequences are added to the MSA.
Instead of just counting the frequency of each amino acid type at a given column in the MSA and dividing by the total number of sequences, HHsearch computes a weighted frequency based on sequence identity (percentage of identical residues between multiple sequences in the alignment).
Suppose a Cysteine residue occurs 5 times across five template sequences with below 10% sequence identity. Suppose that at the same column, Glyceine occurs 5 times across five template sequences that share over 80% sequence identity. Intuitively, the emission probability for Cysteine should be higher than that of Glyceine since multiple occurrences in divergent template sequences are a stronger indication that it contributes to core structures across the protein family than occurrences from similar sequences.
When a residue of type a occurs in a sequence n with low sequence identity to the other sequences in the alignment, we consider it an independent occurrence of a; whereas if sequence n is highly similar to the other sequences in the alignment, we consider it a correlated occurrence of a.
Therefore, we compute the weighted frequencies of each residue type at each column of the MSA as follows:
Since local alignment results in several subdomains of highly aligned fragments, we split the MSA into subdomains and cluster sequences with high sequence similarity at each subdomain. A sequence is added to a cluster if the percentage of identical residues with at least one sequence in the cluster exceeds a specified threshold.
Then, for a column within a specific subdomain of the MSA, the weighted frequency of each amino acid type is computed by counting each occurrence of that amino acid divided by the size of the cluster where the amino acid occurs.
This weighting scheme effectively places more weight on an occurrence originating from sequences in small clusters (independent occurrences) than on occurrences from sequences in large clusters. If a type a residue occurs across most of the sequences in a large cluster, the total count that the cluster contributes to the frequency of type a will be close to 1, whereas if a occurs at only one sequence in a large cluster, this indicates a random mutation and will barely contribute to the count for a after by scaled down by the size of the cluster.
If C_i(n) denotes the cluster containing sequence n at column i of the query MSA, then the weighted frequency of a residue a appearing in column i is given by:
\(\underbrace{\sum_{n=1}^N\frac{\text{count}_{n,i}(a)}{\text{size}(C_i(n))}}_{\text{weighted freq of residue }a\text{ in column }i}\)
where count(a) is equal to 1 if a residue of type a appears at position i of the sequence, and 0 otherwise.
We divide the weighted frequency by the total number of clusters in column i of the MSA to get the probability of observing an independent occurrence of residue a in column i, such that the probabilities across all 20 amino acids sum to 1.
We repeat this process for all 20 amino acid types gives us the set of observed frequencies of each residue type in column i of the query MSA.
\(\underbrace{\{q^{\text{obs}}_i(a)\}_{a=1}^{20}}_{\text{observed probabilities of each amino acid occuring at column }i}\)
Even with the observed frequencies, we still want to retain some information about the known evolutionary mutation patterns as indicated in the substitution matrices. Therefore, we use the observed probabilities to compute an expected probability of finding residue x at column i using the substitution probability that we computed earlier from the Gonnet matrix.
\(\underbrace{q^{\text{exp}}_i(x)}_{\text{expected probability}}=\underbrace{f(x)}_{\text{background freq}}\underbrace{\sum_{a=1}^{20}q_i^{\text{obs}}(a)\cdot g_{ax}}_{\text{weighted probability of replacement with }x}\)
The product inside the summation is the probability of replacing a residue of type a with x weighted by its observed probability in column i. Therefore, the sum is the weighted probability of replacing any residue in column i with a residue of type x.
\(\underbrace{\sum_{a=1}^{20}q_i^{\text{obs}}(a)\cdot g_{ax}}_{\text{weighted probability of replacing the residue in column }i\text{ with }x}\)
Note that since we normalized g_ax such that it sums to 1, the weighted probability will never exceed the maximum observed probability.
We scale this value by f(x) which is the background probability of residue x since the expected probability should be higher for commonly occurring amino acid types in nature.
To combine the observed and expected probabilities into a single emission probability for residue x at column i, we take a weighted average scaled by constants α and β.
In the PSI-BLAST algorithm, α is set to the number of template sequences in the MSA. Therefore, as the number of template sequences increases, the contribution of the expected probability decreases. From experimentation, PSI-BLAST set β = 10.
A similar procedure used to compute observed amino acid probabilities is used to compute the set of transition probabilities from each column in the MSA.
\(q^{\text{obs}}_{i-1}(X_{i-1},X')=\frac{1}{N_\text{clusters}}\sum_{n=1}^N\frac{\text{count}_{n,i}(X')}{\text{size}(C_i(n))}\tag{$X'\in \{M, I, D\}$}\)
After initializing the query HMM profile, we can use it to align new template sequences with the Viterbi algorithm.
Log-Odds Score with Viterbi Algorithm
Thelog-odds score measures the similarity between the query HMM and a template sequence by comparing the probability of the path created by the template sequence through the query HMM to the probability of the same path through a null model that randomly generates amino acids from a background frequency.
Since this will be a math-heavy section, let’s clearly define our notation.
\(\small\begin{align}&l\in[1\dots L] &\text{index for the amino acids in the template sequence}\\&x_l\in \{1,\dots, 20\}&\text{index of amino acid at position $l$ in the alignment}\\&i\in [1\dots N]&\text{index for the match states in the query HMM}\\&q_i(x_l)&\text{probability that column $i$ in the query HMM emits residue $x_l$}\\&f(x_l)&\text{background frequency of residue $x_l$ (same at every position)}\\&M&\text{match state}\\&I&\text{insertion state}\\&D&\text{deletion state}\\&X_i\in \{M,I,D\}&\text{state at column $i$ of the query HMM}\\&q_{i-1}(X_{i-1},X_i)&\text{transition probability from state $X_{i-1}$ to state $X_i$ in query HMM}\end{align}
\)
To determine whether the template sequence is a homolog, we can compute the maximum log-odds (LO) score that measures how much more likely it is for the template sequence to be emitted from the query HMM profile (q) than from a null model that emits residues at their background frequency.
\(\begin{align}S_{\text{LO}}&=\log\left(\frac{P(x_1,...,x_L|\text{emission on path})}{P(x_1,...,x_L|\text{null})}\right)\nonumber\\\\&=\log\left(\frac{\prod_{l=1}^Lq_i(x_l)\cdot\prod_{i=2}^{N}q_{i-1}(X_{i-1}, X_{i})}{\prod_{l=1}^Lf(x_l)}\right)\end{align}\)
The numerator is the probability of a possible path created by the query sequence through the template HMM given by the product of the emission probabilities for each residue in the template sequence x_1, …, x_L multiplied with the product of the transition probabilities between every state along the path, where X ∈ {M, I, D}.
\(P(x_1,...,x_L|\text{emission on path})=\underbrace{\prod_{l=1}^Lq_i(x_l)}_{\text{emission probabilities}}\cdot\underbrace{\prod_{i=2}^{N}q_{i-1}(X_{i-1}, X_{i})}_{\text{transition probabilities}}\)
The denominator is the product of the probabilities of each residue x_l of the template sequence based on the fixed background frequency for the 20 amino acid types f(a), where a ∈ [1, …, 20].
Since the deletion state is not aligned with a residue and the emission probability at an insertion state equals the background probability which cancels in the numerator and denominator, we only multiply the emission probabilities of residues aligned with a match state.
In the figure below, we feed the template sequence CYKYPhD through a specific path of the query HMM and calculate the log-odds score by multiplying the log-ratio emission probabilities of each residue at a match state (C, Y, K, Y, P, and D) and the transition probabilities between all states.
The alignment and log-odds scores given the emission of a template sequence through a specific path in the query HMM profile. (Source: Alchemy Bio)
You may already have noticed that multiple paths through the query HMM can produce the same final sequence (match states can be switched out with insertion states, deletion states can occur between residues, etc.). That is, there are multiple possible ways to align a given template sequence with the query HMM with different LO scores; so we need to find the path corresponding to the best possible alignment.
Consider a query HMM and a template sequence (x_1, …, x_L) indexed from l = 1,…, L.How do we determine which column of the query MSA the first residue x_1 aligns with? What about x_2? Should they be adjacent in the alignment or would adding insertion or deletion positions in between increase the score of the alignment?
The Viterbi algorithm is a recursive algorithm that computes the LO score step-by-step, where each step requires the LO score from the previous steps. To see how this works, let’s break down our LO score equation to isolate the last step leading to the last residue x_L of the template sequence aligned with the last column N of the query HMM from all the previous steps using the product rule oflogarithms1.
\(\begin{align}S_{\text{LO}}&=\log\left(\frac{\prod_{l=1}^Lq_i(x_l)\cdot\prod_{i=2}^{N}q_{i-1}(X_{i-1}, X_{i})}{\prod_{l=1}^Lf(x_l)}\right)\\&=\log\left(\frac{\prod_{l=1}^Lq_i(x_l)}{\prod_{l=1}^Lf(x_l)}\right)+\log\left(\prod_{i=2}^{N}q_{i-1}(X_{i-1}, X_{i})\right)\\&=\underbrace{\log\left(\frac{q_i(x_L)}{f(x_L)}\right)}_{\text{log ratio for residue }x_L}+\underbrace{\sum_{l=1}^{L-1}\log\left(\frac{q(x_L)}{f(x_L)}\right)}_{\text{sum of log ratios for previous steps}}+\underbrace{\log\bigg(q_{N-1}(X_{N-1}, X_{N})\bigg)}_{\text{transition probability to last state}}+\underbrace{\sum_{i=2}^{N-1}\log\left(q_{i-1}(X_{i-1}, X_{i})\right)}_{\text{sum of transition probabilities for previous steps}}\\&=\underbrace{\log\left(\frac{q_i(x_L)}{f(x_L)}\right)+\log\bigg(q_{N-1}(X_{N-1}, X_{N})\bigg)}_{\text{last step}}+\underbrace{\sum_{l=1}^{L-1}\log\left(\frac{q_i(x_L)}{f(x_L)}\right)+\sum_{i=2}^{N-1}\log\left(q_{i-1}(X_{i-1}, X_{i})\right)}_{\text{sum of previous steps}}\end{align}\)
If we denote the last step as S(L, N) and the score up to the previous step as S(L-1, N-1), we see that the LO score becomes a recurrence relation:
We have shown that we can compute the LO one position of the alignment at a time, where at each position, we add a new term to the score of the alignment leading up to the previous position. But since there are three possible states at each column of the query HMM and local alignment allows the first residue x_1 to align with any column, we need an algorithm that computes each score for each residue-column pair with the maximum possible score up to the previous residue-column pair for all possible combinations of states.
\(\begin{align}S_{\text{LO}}(l,i)=\underbrace{\log\left(\frac{q_i(x_l)}{f(x_l)}\right)}_{\text{emission}}+\underbrace{\log\bigg(q_{i-1}(X_{i-1}, X_{i})\bigg)}_{\text{transition}}+\underbrace{\max\bigg(S_{\text{LO}}(l-1, i-1)\bigg)}_{\text{score of best partial alignment path}}\end{align}\)
This emission term of this equation is positive only when the query HMM at column i has a higher probability of emitting residue x_l than the null model.As the emission probability of the query HMM drops below the null model, this term decreases exponentially towards negative infinity.
Therefore, to prevent the negative log-emission ratios of highly non-conserved regions from dominating the LO score for an alignment that starts later along the sequence, let’s introduce local alignment as an alternative to semi-global alignment.
Semi-global alignment aligns the entire template sequence with a column of the query HMM (or query sequence) but local alignment only aligns a subset of columns between the query and template sequences which prevents penalizing unaligned residues for homologs that are only aligned at a core subsequence. (Source: Alchemy Bio)
Semi-global alignment computes the alignment score by aligning every residue in the template sequence with a column of the query profile, meaning that the entire template sequence contributes to the final score determining whether it is classified as a homolog.
In contrast, local alignment can produce high LO scores for alignments where only a subsequence of the template sequence is aligned with a subsequence of columns in the query HMM, even if there are leading or trailing residues that have negative LO scores with the query HMM. In other words, local alignment doesn’t penalize unaligned residues before or after an aligned region in the query or template sequences. Since protein homologs are often characterized by core subsequences that define their primary function, local alignment has been shown to produce much better results than semi-global alignment.
To implement local alignment, instead of just taking the maximum partial alignment score of the previous residue-state pair, we return zero if all partial alignment scores leading up to the previous residue-column pair are negative. This prevents an unaligned region before the sequence of residue-state pairs with positive emission probabilities from lowering the LO score.
\(\begin{align}S_{\text{LO}}(l,i)=\underbrace{\log\left(\frac{q_i(x_l)}{f(x_l)}\right)}_{\text{emission}}+\underbrace{\log\bigg(q_{i-1}(X_{i-1}, X_{i})\bigg)}_{\text{transition}}+\underbrace{\max\bigg(0, S_{\text{LO}}(l-1, i-1)\bigg)}_{\text{if all partial alignment scores are negative, reset to 0}}\end{align}\)
After the recursive algorithm ends, we prevent unaligned regions after a local alignment from negatively contributing to the LO score by taking the maximum intermediate score across all residue-column pairs as the final LO score for the alignment, even if it doesn’t correspond to the last column of the query HMM or the last residue in the template sequence.
Now that we understand the theory behind the LO score, let’s discuss how to store and retrieve the maximum previous alignment at each recursive step using a fundamental data structure of recursive algorithms: dynamic programming matrices (DPMs).
Dynamic Programming Matrices
Dynamic programming matrices (DPMs) are used to store the output of recursive algorithms that recurse on two different variables. Here, we are recursing on the residue in the template sequence x_l and the column in the query HMM i. The DPM is initialized with only the base case values and recursively populated at each recursive step. This data structure provides an efficient way to store and retrieve the results from the previous recursive step.
To store all intermediate values for each recursive call, we must define a dynamic programming matrix (DPM) for each of the three states M, I, and D.
Each DPM is an L x N matrix where a row corresponds to a single residue in the template sequence and a column corresponds to a column or match state in the query HMM.
The (l, i)th entry of the match state DPM represents the partial LO score of the best path leading to an alignment between residue x_l in the template sequence with the ith match state in the query HMM.
The (l, i)th entry of the insertion state DPM represents the partial LO score of the best path leading to an insertion state that emits the residue x_l at the ith column in the query HMM.
The (l, i)th entry in the deletion state DPM represents the partial LO score of the best path leading to a deletion state after residue x_l in the template sequence at the ith column in the query HMM.
Since the goal of local alignment is to test out all possible paths of aligned match states while freely discarding unaligned regions that produce negative log-emission ratios, we initialize the DPMs such that an alignment cannot start or end at an insertion or deletion state so that we compute the score for every possible starting match state.
Initializing the Dynamic Programming Matrix (DPM) of each state for local alignment. (Source: Alchemy Bio)
With this idea in mind, we can initialize each DPM as follows:
The first row of the match state DPM is initialized with the log-likelihood ratio of emitting x_1 at each match state in the query HMM, for columns i = 1, …, N. The first column of the DPM is initialized with the log-likelihood ratio of the first match state emitting any of the residues in the template sequence, for residues l = 1,…, L.
where ‘?’ denotes entries of the DPM that have yet to be filled.
The first row of the insertion state DPM is initialized to negative infinity to prevent the first residue x_1 from being aligned with an insertion state. Since the first column corresponds to the probabilities of the path starting from the first match state aligned with the residue x_1 and all subsequent residues being emitted from insertion states, the entries in the first column are not initialized and will be computed recursively.
In contrast to the insertion DPM, the first column is of the deletion state DPM is initialized to negative infinity to prevent the alignment starting at the first column from being a deletion state and ensuring that a deletion state can only be reached after at least one match state. Since the first row corresponds to the probability of transitioning to deletion states from residue x_1 without emitting the next residue, the entries in the first row are not initialized and will be computed recursively.
Since we need to compute the maximum previous alignment, we can only transition out of a residue-column pair that is defined in all three DPMs. The only residue-column pair we can start the recursion from is the entry (1,1) or the alignment between the first residue x_1 of the template sequence with the first column of the query HMM.
Starting from the first match position aligned with the first residue x_1 (top-left entry of each DPM), we can transition into one of three states: (1) another match state that emits residue x_2 and transitions to the 2nd column in the HMM, (2) an insertion state that emits x_2 according to the background frequency and remains at the 1st column, or (3) a deletion state that does not emit a residue and remains on residue x_1 but transitions to the 2nd column.
When computing the best partial alignment score for the path leading from a previous state at entry (l-1, i-1)to one of the three states X_i ∈ {M, I, D}, we add the log-ratio of the emission probability to the maximum score at entry (l-1, i-1) across the DPMs of all the states that can transition into X_iadded to the transition probability.
But since entry (1, 1) of the insetion and deletion DPMs are initialized to -∞, the maximum score is automatically entry (1, 1) of the match state DPM. Additionally, we know that only the match state has a non-zero emission probability, so the score for the partial path from column (1, 1) to a I or D state are equal to the value at the match state DPM at entry (1, 1) added to the log transition probability.
\(\begin{align}S_M(2,2)&=\underbrace{\log\left(\frac{q_{2}(x_2)}{f(x_2)}\right)}_{\text{emission of residue }x_2}+\max\begin{cases}0\text{ (for local alignment)}\\\underbrace{S_M(1, 1)}_{\text{from initialization}}+\underbrace{\log\big(q_{1}(M,M)\big)}_{\text{transition probability}}\end{cases}\tag{1}\\S_I(2,1)&=\underbrace{\log\left(\frac{f(x_2)}{f(x_2)}\right)}_{=0}+\underbrace{S_M(1, 1)}_{\text{from initialization}}+\underbrace{\log\big(q_{1}(M,I)\big)}_{\text{transition probability}}\tag{2}\\S_D(1,2)&=\underbrace{S_M(1, 1)}_{\text{from initialization}}+\underbrace{\log\big(q_{1}(M,D)\big)}_{\text{transition probability}}\tag{3}\end{align}\)
Computing the LO scores for transitions out of entry (1, 1). Since the insertion and deletion states are -∞ at (1, 1) the recursive step automatically takes the score at (1, 1) of the match state DPM. (Source: Alchemy Bio)
After the first recursive step, there are now two entries defined in all three DPMs: entry (1, 2) and entry (2, 1).
First, let’s consider the three states that can be reached from the residue-column pair (1, 2) corresponding to the alignment between residue x_1 with the 2nd column of the query HMM: (1) a match state that emits residue x_2 and transitions to the 3rd column in the HMM, (2) an insertion state that emits x_2 according to the background frequency and remains at the 2nd column, or (3) a deletion state that does not emit a residue but transitions to the 3rd column.
\(\begin{align}S_M(2, 3)&=\underbrace{\log\left(\frac{q_3(x_2)}{f(x_2)}\right)}_{\text{emission of }x_2 \text{ at 3rd M state}}+\underbrace{\max\begin{cases}0\text{ (for local alignment)}\\S_M(1, 2)+\log\big(q_2(M, M)\big)\\-\infty\\S_D(1, 2)+\log\big(q_2(M, D)\big)\end{cases}}_{\text{best path from alignment (1, 2) to match state}}\tag{1}\\\\S_I(2, 2)&=\underbrace{\max\begin{cases}S_M(1, 2)+\log\big(q_2(M, I)\big)\\-\infty\end{cases}}_{\text{best path from alignment (1, 2) to insertion state}}\tag{2}\\\\S_D(1,3)&=\underbrace{\max\begin{cases}S_M(1, 2)+\log\big(q_2(M, D)\big)\\S_D(1, 2)+\log\big(q_2(D, D)\big)\end{cases}}_{\text{best path from alignment (1, 2) to deletion state}}\tag{3}\end{align}
\)
Notice that there are three terms in the maximization for the M state DPM because all states can transition into a match state. But given that D → I and I → D transitions are not allowed, there are only two terms in the maximization for the I and D state DPMs.
In addition, we can observe that the value at (1, 2) of the insertion DPM is initialized to negative infinity, meaning no path can pass through this entry and we ignore it in the maximization.
Computing the scores for the partial alignments that can be reached from the (1, 2) residue-column pair. (Source: Alchemy Bio)
Next, we can consider the transitions from the residue-column pair (2, 1) corresponding to the alignment between residue x_2 with the 1st column of the query HMM. Similarly, we have three cases: (1) a match state that emits residue x_3 and transitions to the 2nd column in the HMM, (2) an insertion state that emits x_3 according to the background frequency and remains at the 1st column, or (3) a deletion state that does not emit a residue but transitions to the 2nd column.
\(\begin{align}S_M(3, 2)&=\underbrace{\log\left(\frac{q_2(x_3)}{f(x_3)}\right)}_{\text{emission of }x_3 \text{ at 2nd M state}}+\underbrace{\max\begin{cases}0\text{ (for local alignment)}\\S_M(2,1)+\log\big(q_1(M, M)\big)\\S_I(2, 1)+\log\big(q_1(M, I)\big)\\-\infty\end{cases}}_{\text{best path from alignment (2, 1) to match state}}\tag{1}\\\\S_I(3, 1)&=\underbrace{\max\begin{cases}S_M(2, 1)+\log\big(q_1(M, I)\big)\\S_I(2,1)+\log\big(q_1(I, I)\big)\end{cases}}_{\text{best path from alignment (2, 1) to insertion state}}\tag{2}\\\\S_D(2,2)&=\underbrace{\max\begin{cases}S_M(2, 1)+\log\big(q_1(M, D)\big)\\-\infty\end{cases}}_{\text{best path from alignment (2, 1) to deletion state}}\tag{3}\end{align}\)
Here, the value at (2, 1) of the deletion DPM is initialized to negative infinity, so we ignore it in the maximization.
Computing the scores for the partial alignments that can be reached from the (2, 1) residue-column pair. (Source: Alchemy Bio)
From the first two recursive steps, we start to observe a pattern.
When populating the match state DPM, we take steps diagonally towards the bottom-right corner because transitioning to a match state increments both the residue and the column index. The (l, i)th entry is computed by adding the log emission probability of residue x_l at the ith match state to the maximum sum of the top-left adjacent entry at index (l-1, i-1) across the match, insertion, and deletion DPMs and adding the associated transition probability from the previous state to a M state.
When populating the insertion state DPM, we take steps horizontally across each row because transitioning to an insertion state only increments the residue index but not the column index. The (l, i)th entry is computed by taking the maximum sum of the entry directly above at index (l-1, i) in either the insertion state DPM itself or the match state DPM with the transition probability.
When populating the deletion state DPM, we take the recursive steps vertically down each column because transitioning to a deletion state only increments the column index but not the residue index. The (l, i)th entry is computed by taking the maximum sum of the entry directly to its left at index (l, i-1) in either the deletion state DPM itself or the match state DPM with the transition probability.
Since computing each entry of each DPM requires the entries from the state DPMs, we must populate the matrices in parallel, one entry of each matrix at a time.
As we populate each DPM, we simultaneously store the previous state of the best partial alignment used to compute each entry (the state corresponding to the score returned by the maximization) in three backtracking (bt) matrices for each state: M, I, and D.
For instance, if we’re computing the entry (i, j) of the match state DPM and the maximization returns the score from the entry (l-1, i-1) of the insertion DPM, then the (i, j)th entry of the match state backtracking matrix is set to I.
Since the maximization of the match DPM can return 0 for local alignment, the corresponding entry of the bt matrix is set to a stopping state ‘STOP,’ which indicates the start of an alignment.
For the match bt matrix, the entries are determined as follows:
\(\small \text{for }S_M(l, i)\text{ if }\max=\begin{cases}0\text{ (for local alignment)}&\text{bt}_M(i, j)=\text{'STOP'}\\S_M(l-1, i-1)+\log\big(q_{i-1}(M,M)\big)&\text{bt}_M(i, j)=\text{'M'}\\S_I(l-1, i-1)+\log\big(q_{i-1}(I,M)\big)&\text{bt}_M(i, j)=\text{'I'}\\S_D(l-1, i-1)+\log\big(q_{i-1}(D,M)\big)&\text{bt}_M(i, j)=\text{'D'}\end{cases}\)
Once we have populated all L x N entries in all three DPMs and their backtracking matrices, we extract the maximum entry (l_max, i_max) in the match state DPM. The score at(l_max, i_max) will correspond to the final residue-column pair in the best local alignment.
From entry (l_max, i_max) of the match backtracingmatrix, we can follow the back pointers through the backtracking matrices until we reach a ‘STOP’ pointer to determine the sequence of states that determine whether each residue of the template sequence is aligned with a residue, an insertion, or a deletion in the query MSA.
Depending on the state of the back pointer at entry (i, j), we transition to the specified entry of one of the three backtracking matrices. (Source: Alchemy Bio)
If the back pointer at entry (l, i) is ‘M’, we move to entry (l-1, i-1) of the match bt matrix; if it is ‘I’, we move to entry (l, i-1) of the insertion bt matrix; and if it is ‘D’, we move to entry (l-1, i) of the deletion bt matrix.
Below, I have written my version of the Viterbi algorithm for query HMM to template sequence alignment that consolidates all the steps described above.
This algorithm was written to align closely with the notation used for the HMM-HMM alignment algorithm for HHsearch so that you can clearly see how the steps for HMM-sequence alignments translate into HMM-HMM alignment in the next section.
Viterbi algorithm for query HMM and template sequence alignment. (Source: Alchemy Bio)
The maximization function MAX3 returns the maximum partial alignment score across the match, insertion, and deletion DPMs and the associated state to store in the backtracking matrix.
A function that returns the maximum partial alignment score and the previous state for backtracing. (Source: Alchemy Bio)
With the intuition from computing the log-odds score for sequence-HMM alignments, let’s level up to HMM-HMM alignments and compute the log-sum-of-odds score.
Log-Sum-Of-Odds Score
The log-sum-of-odds score extends the log-odds score HMM-HMM alignment by measuring the similarity between the query HMM and a template HMM. It is the log-probability ratio of both HMMs co-emit any given sequence of amino acids compared to a null model.
Here’s a reminder of our notation with some symbols specific to the LSO score:
\(\scriptsize\begin{align}&l\in[1\dots L] &\text{index for the amino acids in the template sequence}\\&x_l\in \{1,\dots, 20\}&\text{index of amino acid at position $l$ in the alignment}\\&i\in [1\dots N_q]&\text{index for the match states in the query HMM}\\&j\in [1\dots N_t]&\text{index for the match states in the template HMM}\\&q_i(x_l)&\text{probability that column $i$ in the query HMM emits residue $x_l$}\\&t_j(x_l)&\text{probability that column $j$ in the query HMM emits residue $x_l$}\\&f(x_l)&\text{background frequency of residue $x_l$ (same at every position)}\\&X_i,Y_j\in \{M,I,D\}&\text{states at column $i$ and column $j$ of the query and template HMM respectively}\\&q_{i-1}(X_{i-1},X_i)&\text{transition probability from state $X_{i-1}$ to state $X_i$ in query HMM}\\&t_{j-1}(Y_{j-1},Y_{j})&\text{transition probability from state $Y_{j-1}$ to state $Y_j$ in template HMM}\\&\mathcal{P}_{tr}&\text{short form for product of transition probabilities between pair states}\end{align}
\)
To increase alignment sensitivity further, HHsearch introduced an HMM-HMM search algorithm that computes the score of the best local alignment between the query HMM profile and a template HMM profile by computing the highest Log-Sum-Of-Odds (LSO) score across all possible sequences of pair-alignment states.
To compute the log-sum-of-odds score for a single alignment between the query and template HMM, all possible sequences of length L are fed into both the query and template HMM profiles to compute the log-likelihood that any sequence is co-emitted along the paths defined by a series of pair alignment states. (Source: Alchemy Bio)
The LSO score is an extension of the LO score that measures the log-likelihood that a specific series of pair alignment states between the query HMM q and template HMM t will co-emit any given sequence of amino acids with length L (length of the query sequence).
\(S_{\text{LSO}}=\underbrace{\log\left(\frac{P(x_1,...,x_L|\text{co-emission on paths})}{P(x_1,...,x_L|\text{null})}\right)}_{\text{log-likelihood of co-emitting any given sequence given a path through both HMMs}}\)
For sequence-HMM alignments, we determined the path through the query HMM that produces the best log-odds score when emitting a single sequence.
But for HMM-HMM alignments, we need to determine the pair of aligned paths through the query and template HMMs that has the highest log-likelihood of co-emitting the same sequence, no matter the identity of the sequence itself.
To compute how well both HMMs emit the same sequence for a given alignment, we align a path through the query HMM with a path through the template HMM and compute how likely both paths emit the same residue sequence in parallel. We call this a co-emission path.
To do this, we define the set of pair alignment states of the form XY that defines the state of the query HMM X that is aligned with the state of the template HMM Y in the co-emission path. The series of XY states with the highest score determines the specific positions of the query and template sequences that will be aligned in the final MSA.
A Match-Match (MM) state occurs when the amino acid at position l is emitted as a conserved residue by both the query HMM and the template HMM in the co-emission path. The co-emission probability is the product of the query emission probability at column i and the template emission probability at column j, where each is generated from the amino acid frequencies used to construct the HMM.
A Match-Insertion (MI) state occurs when the amino acid at position l is emitted as a conserved residue by the query HMM and a variable residue by the template HMM in the co-emission path. The co-emission probability is the product of the query emission probability and the background frequency. Since insert states exist between two adjacent match states in an HMM profile, the column index j of the template HMM is not incremented when transitioning from the previous state to the insertion state.
Intuitively, a MI state occurs when a pair of non-adjacent columns separated by variable positions in the template sequence is aligned with a pair of adjacent columns in the query sequence.
An Insertion-Match (IM) state is the inverse of the MI state and occurs when the amino acid at position l is emitted as a variable residue by the query HMM and a conserved residue by the template HMM in the co-emission path. The co-emission and transition probabilities are similar to the MI state except the column index of i of the query HMM is not incremented.
A Deletion-Gap (DG) occurs when the query HMM transitions to a deletion state at column i without an aligned state in the template HMM, which we denote with a gap state. Since both the deletion and gap states do not emit amino acids, we set the co-emission probability to 1. There is a transition probability associated with the transition into the deletion state in the query HMM but since the gap state is not associated with a state in the template HMM, it does not have a transition probability, and the column of the template HMMis not incremented.
Intuitively, a DG state occurs when a pair of adjacent columns separated by a high density of deletions in the query sequence is aligned with an pair of adjacent columns that are conserved in the template sequence.
A Gap-Deletion (GD) state inverse of the DG state and occurs when the template HMM transitions to a deletion state at column j without an aligned state in the query HMM, which we denote with a gap state.
From a series of pair alignment states, we can derive the MSA alignment of the template sequence with the query sequence, where the positions j in the template sequence associated with MM pair states are aligned in the same column as the matched position i in the query MSA.
Before moving on, there are a few things to note.
First, switching an MI state for a DG or a IM state for a GD state generates the same alignment, but we distinguish them because the transition probabilities differ depending on the frequency of insertion or deletion states in the original sequences.
Second, to prevent unessesary insertion and deletions along the alignment, we exclude II and DD pair alignment states which can be replaced with MM states.
Allowed transitions between pair-alignment states. (Source)
Finally, for a similar reason to our second point, we only consider transitions between the MM state all remaining states, as well as transitions between a state and itself.
Similar to the LO score, the LSO score for a specific alignment of the query and template HMMs measures how much more likely it is for a sequence to be co-emitted along the series of pair alignment states throughthe query HMM and the template HMMcompared to being emitted from a null model.
But since we are comparing two profiles without a defined sequence, we have to sum over the probability ratios for all possible sequences with length L(x_1, …, x_L) of the 20 types of amino acids to compute the total LSO score for the alignment.
\(\begin{align}S_{\text{LSO}}&=\log\underbrace{\sum_{x_1=1}^{20}\dots\sum_{x_L=1}^{20}\underbrace{\left(\frac{\prod_{l=1}^Lq_i(x_l)t_j(x_l)\times \mathcal{P}_{tr}}{\prod_{l=1}^Lf(x_l)}\right)}_{\text{probability of co-emission along paths divided by null model}}}_{\text{sum over all possible sequences of length }L}\end{align}\)
Similarly to sequence-to-HMM alignments, there are multiple possible alignments of the columns of the query and template HMM corresponding to different co-emission paths, so we need to determine the path that gives the highest LSO score with the Viterbi algorithm.
To do this, let’s convert the equation for the LSO score into a form that can be computed recursively:
Since the transition probabilities are not dependent on the amino acids that are emitted, we take it out of the summation and expand it into its full form.
\(\small S_{\text{LSO}}=\log\underbrace{\sum_{x_1=1}^{20}\dots \sum_{x_L=1}^{20}\prod_{l=1}^L\left(\frac{q_i(x_l)t_j(x_l)}{f(x_l)}\right)}_{\text{sum of co-emission probabilities for all sequences}}\times\log\underbrace{\prod_{ij}\bigg(q_{i-1}(X_{i-1},X_{i})t_{j-1}(Y_{i-1},Y_{i})\bigg)}_{\text{product of all pair state transition probabilities}}\)
In the above equation, we are taking the sum of the co-emission probability for all possible sequences of length L through the pair alignment path. By observing that the emission probabilities of each residue are mutually independent across each sequence, we can rearrange this equation to compute the log-emission ratio of q and t co-emitthing any oneof the 20 amino acid residues for each pair of aligned columns i and j.
Now, we have the final form of the LSO score for recursive computations:
\(S_{\text{LSO}}=\sum_{ij}\underbrace{S_{aa}\big(q_{i}(a),t_{j}(a)\big)}_{\text{column score}}+\underbrace{\sum_{ij}\log\bigg(q_{i-1}(X_{i-1},X_{i})t_{j-1}(Y_{i-1},Y_{i})\bigg)}_{\text{sum of all pair state transition probabilities}}\)
To gain some intuition as to why the equation above finds the best local alignment between HMMs, let’s break down the column score.
Column Score
The column score, Saa(q_i, t_j), is the likelihood that the ith column of the query HMM profile and the jth column of the template HMM profile are homologous pairs. In other words, the column score is high when a pair of columns are conserved in the query and template MSA and emit similar amino acid profiles.
The numerator is the co-emission probability that both the ith column of the query HMM and the jth column of the template HMM emit the amino acid a.
The denominator is the background frequency of a, f(a), which is the percentage in which a appears in a normal protein sequence. Intuitively, dividing by f(a) scales the probability of alignment to reflect the likelihood of it occurring by chance rather than by homologous alignment.
For amino acids that appear often in natural proteins (larger f(a) value), the probability that both HMMs emit a by chance is high, thus an alignment involving a has a less significant contribution to the total score. For rare amino acids (smaller f(a) value), the probability that both HMMs emit a by chance is low, thus an alignment involving a has a greater contribution to the total score.
To understand why this equation works, let’s consider the following cases.
If a pair of aligned columns in the query and template HMMs are conserved positions with similar amino acid emission profiles, the column score will be positive and increase the total LSO score.
Consider when column i of the query HMM is perfectly conserved and emits a single amino acid a with a probability of 1 and all other amino acids with probabilities of 0. Then, the q_i(a) term in the numerator disappears, resulting in a column score that is only dependent on the probability that the aligned column j of the template sequence emits the same amino acid a.
When the pair of aligned query and template are non-homologous, the column score will be negative and decrease the total LSO score. Two columns are considered non-homologous if, for any amino acid a, the combined probability that both the query and template profiles emit a is less than or equal to the background frequency. In this case, the probability ratio is less than 1, and the log ratio is negative.
Consider the case where the query profile at column i is perfectly conserved and emits Cysteine with a probability of 1 and all other residues at a probability of 0 and the template profile at column j is also perfectly conserved and emits Glycine with a probability of 0.99 and Cysteine with a probability of 0.01. Therefore, for all residues a, product of the probabilities that the two profiles emit a will be 0 except Cysteine for which it evaluates to 1*0.01 and the resulting column score is negative:
When either the query or template column is in a variable position, where the emission profile closely aligns with the background probabilities f(a), the column score will approach 0 and insignificantly influence the total LSO score.
To illustrate, consider the case when column i of the query HMM emits each type of amino acid at the exact same probability as the background frequency, f(a) will cancel, leaving only the summation of all the emission probabilities for the template sequence in the log function. Since the probability of emitting any amino acid is equal to 1, the column score vanishes to 0.
Since insertion states are variable positions with emission probabilities equal to the background frequency, the column score for MI and IM states are always 0 and we can reduce the LSO score to include only the sum of the emission probabilities of MM pair alignment states.
\(S_{\text{LSO}}=\underbrace{\sum_{ij: X_iY_j=MM}\underbrace{S_{aa}\big(q_{i}(a),t_{j}(a)\big)}_{\text{column score}}}_{\text{sum over MM pair states}}+\log\underbrace{\prod_{ij}\bigg(q_{i-1}(X_{i-1},X_{i})t_j(Y_{j-1},Y_{j})\bigg)}_{\text{product of all pair state transition probabilities}}\)
Viterbi Algorithm for HMM-HMM Alignment
To find the best series of pair alignment states between the query and template HMM, we recursively compute the partial LSO scores for every path using five dynamic programming matrices for each pair alignment state.
Similarly to the LO score, we recursively compute the maximum score leading up to each position (i, j) at each pair alignment state from the maximum partial alignment score from a previous position across the five dynamic programming matrices for each of the pair alignment states (MM, MI, IM, DG, GD).
Instead of adding the log-emission ratio of a specific amino acid like the LO score, we add the column score indicating the log-ratio of both HMMs co-emitting the same amino acid at the aligned columns i and j at each recursive step.
Initialization of pair alignment state DPMs such that all alignments start at the MM pair state. (Source: Alchemy Bio)
Each DPM is N_q x N_t matrix where N_q is the number of columns in the query HMM and N_t is the number of columns in the template HMM. The initialization of each DPM is similar to the single-state DPMs, with a few key differences:
The first row of the MM DPM is initialized with the column scores of the first match state of the query HMM aligned with every match state of the template HMM. Conversely, the first column is initialized with the column scores of the first match state of the template HMM aligned with every match state of the query HMM.
For local alignment, we want every alignment to begin at an MM pair state, so we forbid any alignment from starting at any of the other pair states. From the initial MM state (1, 1), we can transition to an MI or DG to any state (i, 1) without incrementing the column of the template sequence. Therefore, we only initialize the first row of the MI and DG DPMs to negative infinity, preventing an alignment from starting at an MI or DG state.
Conversely, from the initial MM state (1, 1), we can transition to an IM or GD to any state (1, j) without incrementing the column of the query sequence. Therefore, we only initialize the first column of the IM and GD DPMs to negative infinity.
The (i, j)th entry of the DPM for pair state XY stores the LSO score of the best partial alignment of the path up to column i of the query HMM and column j of the template HMM with pair state XY. For the MM pair state, the recursive step takes the maximum across all five DPMs including 0 for local alignment.
Recursive relation for the log-sum-of-odds score for the MM pair state. (Source: Alchemy Bio)
We can break down this recursive relation into three components:
Column Score. The first term is the column score for column i of the query HMM aligned to column j of the template HMM. As discussed earlier, this score is positive for conserved and homologous alignments, 0 for variable alignments, and negative for non-homologous alignments.
Recursion on Maximum Partial Score. The second term is the maximum partial LSO score up to the (i-1, j-1)th entry across all five DPMs added to the log transition probability from the previous pair state (MM, MI, IM, DG, or GD) to state MM.
\(\underbrace{S_{MM}(i − 1, j − 1)}_{\text{partial LSO score up to previous MM state}} + \log\underbrace{\bigg(q_{i−1}(M, M)p_{j−1}(M, M)\bigg)}_{\text{transition prob from MM to MM pair states}}\)
Local Alignment. When the maximum partial LSO score is negative—meaning that the sum of the column scores of the best partial alignment is negative—the maximization returns 0 and we compute the LSO score solely from the column score at (i, j). This allows the alignment to start at any pair of aligned columns without penalty.
Recursive calculation of entry (3, 3) of the Match-Match dynamic programming matrix (DPM) from the maximum partial alignment scores at entry (2, 2) across all five DPMs including itself. (Source: Alchemy Bio)
For the remaining pair states, the only states that transition into it include the MM state and itself, so the maximization reduces to only two states. Furthermore, since the column score of MI and IM states are 0 and the DG and GD states do not emit amino acids, the only term is the maximum partial alignment score and the corresponding transition probability.
For the Match-Insertion (MI) and Insertion-Match (IM) DPMs, we take the maximum entry in the DPMs directly above or to the left of entry (i, j) since transitioning to an insertion state does not increment the column of the HMM.
Transitioning to a gap state does not increment the column of the HMM, so the Gap-Deletion (GD) and Deletion-Gap (DG) DPMs are computed similarly to the MI and IM states respectively. However since the gap state occurs between two adjacent match states in the HMM, the log-transition probability only corresponds to transitioning into the deletion state.
After all five DPMs have iteratively populated, we determine the LSO score of the best local alignment by finding the maximum value in the MM DPM and following the backtracking matrices to a ‘STOP’ state.
Below, I have shown the full Viterbi algorithm for query HMM to template HMM alignment from HHsearch slightly modified for consistency in notation.
As you can see, this algorithm has many similarities to the algorithm for query HMM to sequence alignment except for a few key differences like adding the column score, computing transition probabilities from both HMMs, and storing five DPMs instead of three.
Viterbi algorithm for aligning the Hidden Markov Model profiles of the query sequence and a template sequence. (Modified from Source)
The maximization functions for taking the maximization across five partial LSO scores for the MM DPM and two partial LSO scores are defined below. In MAX2, since we reuse the same function for several different combinations of states, SELF is the back pointer indicating that the pair state remains unchanged from the previous pair state.
The functions that return the maximum partial alignment score and the previous state for backtracing. (Modified from Source)
You may have noticed an unfamiliar ‘structure score’ that was added to each LSO score at an MM state. This score is called the secondary structure score, which we will discuss next.
Secondary Structure Score
A secondary structure score is added to the partial LSO score at each MM pair alignment state that measures the correlation between the predicted secondary structure of a column in the query sequence and the known or predicted secondary structure of a column in the template sequence.
To increase the sensitivity for distant homologs without direct residue alignments, we incorporate a secondary structure score that measures the similarity between the predicted secondary structure (SS) state of a residue in query HMM with the true or predicted SS state of the aligned residue in the template HMM at a HMM pair state.
There are two cases to consider when computing the secondary structure score: when the structure of the template MSA is known and when it is unknown.
We will start with the case when the template SS is known.
The known secondary structure is categorized into one of seven states defined by the DSSP protein database: alpha-helix (H), extended beta-sheet (E), coil (C), 310-helix (G), beta bridge (B), bend (S), and helix-turn (T).
\(\underbrace{\sigma\in\{H, E, C, G, B, S, T\}}_{\text{known secondary structure states}}\)
The predicted SS of a sequence without known structure is generated from the PSIPRED algorithm, which takes a protein sequence as input and predicts the secondary structure of each residue as one of three states: alpha-helix (H), extended beta-sheet (E), and coil (C).
PSIPRED algorithm for residue-wise secondary structure prediction. First, the raw position-specific scoring matrix is normalized into probabilities and fed into a two-network neural network. The network predicts the secondary structure state for each position in the sequence from a window of 15 residues centered at that position. (Source)
From an input sequence, PSIPRED generates an L (length of sequence) x 20position-specific scoring matrix using a substitution matrix method described in the section on initializing the emission probabilities. For each position i, the algorithm feeds a window of 15 residues centered at i through a simple neural network to generate an estimated probability for each of the three SS states. The SS state with the highest probability becomes the final prediction with a confidence score between 0 and 9.
\(\underbrace{\rho\in \{H, E, C\}}_{\text{predicted secondary structure}}\;\;\;\;\underbrace{c\in \{0,\dots9\}}_{\text{confidence score}}\)
Given the known SS state σ of residue j of the template sequence and the predicted SS state ρ and confidence c of residue i of the query sequence, we can compute the secondary structure score with the following equation:
The numerator is the joint probability that the template residue with known secondary structure σ is predicted to have structure ρ with confidence c when fed into the PSPIRED algorithm, where ρ and c are generated from the aligned query residue. Here, the term joint refers to the two events occurring for the same residue in the same protein.
\(\underbrace{P(\sigma;\rho, c)}_{\text{joint probability that a protein with known structure }\sigma\text{ is predicted to have structure }\rho\text{ with confidence }c}\)
If this probability is close to 1, this means that the query residue has similar SS as the template residue such that there is a high chance for the prediction to have been made for the template residue itself. If the probability is close to 0, then this means that the prediction for the query residue is unlikely to be made for the template residue and the two secondary structures are unrelated.
The denominator is the probability that the secondary structure σ and prediction-confidence pair (ρ, c) occur independently from each other for a random pair of residues with no structural correlation. This normalizes the joint probability by a background probability of the two events occurring simultaneously by chance.
\(\underbrace{P(\sigma)P(\rho, c)}_{\text{background probability that the structure }\sigma\text{ and the prediction }(\rho,c)\text{ occur simultaneously by chance}}\)
This probability is higher for common predicted and known SS states where the two are likely to occur together randomly and lower for rare SS states where it is highly unlikely for the two SS states to occur from two random protein residues with no structural correlation.
When the joint probability is much higher than the chance probability, the ratio is greater than 1, yielding a positive log score that increases the total LSO score. When the joint probability is less than the chance probability, the ratio is less than 1, yielding a negative log score that decreases the total LSO score. When the joint probability is exactly equal to the chance probability, the ratio equals 1 and the log score is 0, which doesn’t affect the total LSO score.
These probabilities are generated by feeding all protein domains in the SCOP database into PSIPRED and computing the joint frequency of (σ; ρ, c) and independent frequencies of σ and (ρ, c).
For each confidence score c, the pre-computed SS scores are stored in a 7 x 3 matrix where each row corresponds to a known SS state σ and each column corresponds to a predicted SS state ρ. This value is retrieved and scaled with an empirically determined constant for every alignment between the query and template HMMs.
\(\underbrace{S_{SS}(q_i, t_j)}_{\text{SS score for columns }i\text{ and }j} =0.15\cdot M_{SS}(\sigma^t_j; \rho ^q_i, c^q_i)\)
If the SS state of the template sequence is unknown, then we compute a log-odds score for the predicted SS of the template sequence with the predicted SS of the query sequence.
Computing the secondary structure score when the true SS state for the template and query sequences are unknown. (Source: Alchemy Bio)
Given the SS state and confidence pair predicted by PSIPRED for each residue in both the query and template sequences, we can compute the SS score as:
The numerator is the joint probability that the predicted SS states (ρ, c) of the template and query sequences are derived from an alignment of two homologous sequences that share a true SS state σ.
The denominator is the product of the independent probabilities that both predicted SS states occur simultaneously for any random pair of residues.
Just like the score for known template structure, the log-odds score is positive when the joint probability is higher than the independent probability of the predicted SS states, 0 when they are equal, and negative when the joint probability is lower.
To compute the joint probability, we need to rewrite the numerator as the conditional probability that the predicted SS states(ρ, c) of the template and query sequences occur given a shared secondary structure σ and take the sum over all the possible SS states for σ.
\(P(\rho_i^q,c_i^q;\rho_j^t,c_j^t)=\underbrace{\sum_{\sigma}\underbrace{P(\rho_i^q,c_i^q|\sigma)P(\rho_j^t,c_j^t|\sigma)}_{\text{probability of both predicted states given true SS }\sigma}P(\sigma)}_{\text{sum over all possible true states }\sigma\in\{H, E, C, G, B, S, T\}}\)
The conditional probabilities are obtained from the frequency of prediction (ρ, c)occuring in the subset of domains with true SS state σ.
Substituting this into the SS score equation, we get:
This score measures the likelihood that the predicted SS state and confidence pair (ρ, c) of the template and query sequences to occur from a homologous alignment rather than a random alignment from two sequences without structural dependence.
We scale this by a constant to get the final SS score for column i of the query HMM aligned with column j in the template HMM without known SS states.
\(\underbrace{S_{SS}(q_i, t_j)}_{\text{SS score for columns }i\text{ and }j} =0.15\cdot M_{\text{SS}}(\rho_i^q,c_i^q;\rho_j^t,c_j^t)\)
The SS score is added to the partial LSO score for the MM pair state at every recursive step of the Viterbi algorithm using the residue at position i of the query sequence and position j of the seed sequence of the template HMM.
Correlation Score
Homologous alignments often occur in clusters that define core substructures across the protein family. To further distinguish true homologs from non-homologs with chance alignments, a correlation score is added to the best LSO score for each template sequence.
Since clusters of adjacent residues often form critical substructures that make up the tertiary structure, it follows that conserved subsequences across homologs also occur in clusters.
Therefore, HHsearch incorporates a correlation score that is higher for sequences with high column scores in clusters along the sequence and lower for sequences with the same frequency of high column scores that are scattered along the sequence.
\(g(d)=\underbrace{\sum_{ij}\underbrace{S_{aa}(q_{i}, t_{j})}_{\text{column score at position }ij}\cdot \underbrace{S_{aa}(q_{i+d}, t_{j+d})}_{\text{column score at position }(i+d)(j+d)}}_{\text{sum over all match MM states in alignment}}\)
This equation takes the product of the column score for the alignment between positions i and j and the column score for the position a distance d away in both HMMs. The product inside the summation is 0 if either one of the positions is not an MM state and a positive value between [0, 1] if both positions are MM states.
By taking the sum of the products for every pair of column scores along the best profile-profile alignment with a small separation of d, we generate a score g(d) that indicates the frequency and strength of alignments in nearby positions.
For d = 1, g(1) is computed as the product of column scores between adjacent pairs of alignments along the entire sequence. For d = 2, g(2) is computed as the product of column scores between pairs of alignments with a single position in between. Since clusters can occur within longer stretches of positions and are not limited to adjacent positions, we compute the sum of the g(d) scores for separations up to 4 (d = 1, …, 4). The score is then scaled by an empirically determined constant of 0.1.
\(S_{\text{corr}}=0.1\underbrace{\sum_{d=1}^4g(d)}_{\text{clusters within distance }d}\)
By adding this correlation score to the LSO score for the best alignment of each template sequence, the algorithm can distinguish true homologs with strong structural conservation from sequences with non-homologous alignments that don’t contribute to critical protein substructures.
SIMD and Vectorized Viterbi Algorithm
The newest version of HHsearch introduced with HHsuite-3 introduces several techniques to speed up computation and reduce memory including parallelized computation of the column score for multiple template sequences using single-instruction multiple-data (SIMD) instructions and a vectorized version of the Viterbi algorithm.
Using the original HHsearch algorithm, we need to recursively compute five N_q x N_t DPMs for each template sequence, which requires significant computation time and memory. To increase the practicality of the HHsearch algorithm for alignment against large protein databases without sacrificing the sensitivity of HMM-HMM alignment, various techniques to parallelize the computations were introduced in HHsuite-3.
The accelerated HHsearch algorithm leverages Single-Instruction Multiple-Data (SIMD) programming that enables parallelized processing of multiple pieces of data with a single instruction for tasks involving large databases.
In particular, HHsuite-3 leverages SIMD instructions provided by Streaming SIMD Extensions 2 (SSE2)Advanced Vector Extensions 2 (AVX2) processors. Without going into too much detail on the hardware, SSE2 can process 4 floating-point operations simultaneously with a register that stores 128 bits, which can be used to align 4 template HMMs with a query HMM in parallel. AVX2 can process 8 floating-point operations simultaneously with a register that stores 256 bits, which can be used to align 8 template HMMs in parallel.
At a high level, SIMD can be introduced into algorithms by (1) converting algorithms from scalar operations which process a single data point at a time to vectorized operations that process entire vectors of data at a time and (2) rearranging data into consecutive vectors or matrices in memory so that they can be accessed by the SIMD instructions.
HHsuite-3 leverages SIMD instructions to implement multiple computational speedups, including:
Aligning a single query HMM to 4 or 8 template HMMs in parallel with a vectorized Viterbi algorithm. To compute the LSO scores for 4 (SSE2) or 8 (AVX2) template sequences, we arrange the emission probabilities and transition probabilities consecutively in memory. This allows us to compute the column scores and add the transition probabilities for 4 or 8 template HMM alignments in parallel by loading into a single SIMD register and calling an SIMD instruction.
Reducing storage of dynamic programming matrices. Instead of storing all the full DPM matrix for each pair state, a single vector per pair state stores the partial LSO scores within the same row i as the score (i, j) currently being computed and the scores from the previous row i-1 in the remaining entries.
Reducing storage of five backtracing matrices into a single matrix. Since all backtracking information across each pair state is required to derive the best alignment from the maximum LSO score, we condense all backtracing information into a single N_q x N_t matrix where each entry is a single byte (8 bits).
The previous pair alignment states of the best alignment path are stored as 4-bit (single byte) values in the backtracing matrix. The cell-off bit indicates when a cell should not be passed through. (Source)
There are only two possible back pointers for the pair states MI, IM, DG, and GD: the same or the MM state. Therefore, we store the back pointer for each of the four states as a single bit with 0 signifying a transition to the MM state and 1 signifying a transition into the same previous state. For the MM state, there are 6 possible backpointers: MM, MI, IM, DG, GD, as well as a STOP state. We store this in 3 bits as an integer between 0 (STOP) and 5. The last bit stores a cell-off state that indicates whether the cell can be used in the alignment.
SIMD-based redundancy filter. To avoid aligning redundant template sequences that provide no new information to the MSA, HHsuite removes sequences with greater than a maximum sequence identity (around 70%).
To gain some intuition on how SMID instructions work, we will only be breaking down the vectorized maximization function. To see the implementations of all the remaining speedups using SIMD, refer to the original paper here.
First, we arrange the emission probabilities and transition probabilities consecutively in memory such that for each position, the emission probabilities of a single amino acid type (e.g. Cysteine) and the transition probabilities for a single pair state (e.g. MM) occur in 4 or 8 consecutive locations.
In the Single-Instruction Single-Data (SISD) scalar Viterbi algorithm, the emission probabilities for every amino acid type (columns) and every column of the template HMM (rows) are stored in an N_t x 20 matrix of memory (top). In the single-instruction multiple-data (SIMD) version of the Viterbi algorithm, the emission probabilities for each amino acid type and each position are stored in groups of four template sequences (indicated by the same color) consecutively in memory so that they can be retrieved simultaneously. (Source)
Now, we can implement the VMAX6 algorithm used in HHsuite-3 which takes the vector of partial alignment scores for the current pair state XY (and transition probability) and the previous best partial alignment score stored in S_MM(i, j) and compares the two vectors, updating the elements of S_MM(i, j) and the corresponding index of the backtracing matrix when the alignment score of the current pair state is greater than the previous maximum.
After calling this function five times for all five pair alignment states, the value stored in S_MM(i, j) at each entry will correspond to the maximum partial alignment score for cell (i, j) for the corresponding template HMM.
Vectorized maximization function that is called a total of five times for each of the pair states, updating the partial LSO score stored in S_MM if the one being compared exceeds the previously stored score for each template sequence. The function takes a 4- or 8-dimensional vector containing the scores for multiple template sequences and uses SMID instructions for parallelized computation. (Annotated from Source)
To gain intuition into how this vectorized bit-wise maximization function works, let’s consider the scenario where we are processing 4 template sequences at once and calculating the maximum partial alignment leading to an MM pair state at columns i and j where the first template HMM transitions from an IM pair state (stored as the integer 3 in the back pointer) and all other templates HMM transition from another MM state (stored as integer 1).
First, we define the 4-dimensional vector storing the current partial LSO scores from the first two maximization calls and the vector of scores LSO scores for the IM → MM transition for each template HMM.
\(\small S_{MM}(i, j)=\underbrace{\begin{bmatrix}S^{(1)}_{MM}(i-1, j-1)+\log\left(q_{i-1}(M, M)t^{(1)}_{i-1}(M, M)\right)\\S^{(2)}_{MM}(i-1, j-1)+\log\left(q_{i-1}(M, M)t^{(2)}_{i-1}(M, M)\right)\\S^{(3)}_{MM}(i-1, j-1)+\log\left(q_{i-1}(M, M)t^{(3)}_{i-1}(M, M)\right)\\S^{(4)}_{MM}(i-1, j-1)+\log\left(q_{i-1}(M, M)t^{(4)}_{i-1}(M, M)\right)\end{bmatrix}}_{\text{partial alignment scores are currently set to the MM to MM transition}}\;\;\; S_{IM\to MM}\underbrace{\begin{bmatrix}S^{(1)}_{IM}(i-1, j-1)+\log\left(q_{i-1}(I, M)t^{(1)}_{i-1}(M, M)\right)\\S^{(2)}_{IM}(i-1, j-1)+\log\left(q_{i-1}(I, M)t^{(2)}_{i-1}(M, M)\right)\\S^{(3)}_{IM}(i-1, j-1)+\log\left(q_{i-1}(I, M)t^{(3)}_{i-1}(M, M)\right)\\S^{(4)}_{IM}(i-1, j-1)+\log\left(q_{i-1}(I, M)t^{(4)}_{i-1}(M, M)\right)\end{bmatrix}}_{\text{partial alignment scores from pair state IM to MM for four template sequences}}\)
In the first line of the algorithm, we use the SMID instruction for greater than (gt), which compares the vector of 4 or 8 partial alignment score for a pair state MM to that of the other pair state XY. For each 32-bit entry of the vector, this line outputs 0xFFFFFFFF (32-bits of 1s) when the partial score for MM is greater than that of XY and 0x00000000 when the partial score is less than or equal to MM.
Values where all the bits are 1s or 0s are also called bit-masks because when taking the bit-wise AND operation with another 32-bit value, it will return the value itself if the mask contains only 1s and 0 otherwise.
In line 2, we use the bitwiseAND operation that returns the index of the IM pair state when the bit-mask is 0xFFFFFFFF and 0 if the bit-mask is 0x0000000.
\(\small\text{update_bt}_{i,j}\gets\underbrace{\underbrace{\begin{bmatrix}\text{0x00000003}\\\text{0x00000003}\\\text{0x00000003}\\\text{0x00000003}\end{bmatrix}}_{\text{32-bit index of IM for backtracking}}\&\underbrace{\begin{bmatrix}\text{0xFFFFFFFF}\\\text{0x00000000}\\\text{0x00000000}\\\text{0x00000000}\end{bmatrix}}_{\text{bit-mask}}}_{\text{bit-wise AND}}=\begin{bmatrix}\text{0x00000003}\\\text{0x00000000}\\\text{0x00000000}\\\text{0x00000000}\end{bmatrix}\)
Next, we take the max of the original vector backtracking indices (originally set to 1 across all templates for the MM) and the vector with the index 3 corresponding to the first template that transitions from the IM state to get the updated vector of back pointers.
\(\text{bt}_{i,j}\gets\text{max}\left(\underbrace{\begin{bmatrix}\text{0b00000001}\\\text{0b00000001}\\\text{0b00000001}\\\text{0b00000001}\end{bmatrix}}_{\text{all bt's originally set to MM}},\underbrace{\begin{bmatrix}\text{0b00000003}\\\text{0b00000000}\\\text{0b00000000}\\\text{0b00000000}\end{bmatrix}}_{\text{update first bt to IM}}\right)=\begin{bmatrix}\text{0b00000003}\\\text{0b00000001}\\\text{0b00000001}\\\text{0b00000001}\end{bmatrix}\)
Finally, we use the max operation again to update the actual scores stored in the vector of partial LSO scores.
VMAX6 is called a total of five times to find the maximum partial LSO score across all five pair states that transition into the MM state. The partial LSO scores stored in the S_MM(i, j) are added to a vector of column scores and a vector of secondary structure scores to get the final alignment scores up to the (i, j)th alignment across all 4 or 8 template HMMs.
A similar VMAX2 function is defined to compute the maximum partial LSO score for the remaining pair states MI, IM, DG, and GD and store a back pointer value of either 0 or 1 in the corresponding bit. For the full implementation of VMAX2 and the vectorized Viterbi algorithm, see Algorithm 3 in the original paper.
Iterative MSA Alignment
RF uses an iterative protein sequence searching algorithm called HHblits to first search the large UniRef30 and BFD sequence databases for sequences with aligned local motifs to construct a MSA. Then, the MSA is passed through to HMM-HMM alignment with HHsearch to find template sequences with known structure from the PBD100 database.
Even with the computational speedups, HHsearch is too slow for iterative homology detection for large sequence databases with millions of sequences like UniProt. HHblits is an extension of the HHsearch algorithm that first reduces the pool of sequences using a two-stage prefilter before applying HMM-HMM alignment.
HHblits MSA alignment algorithm that reduces the large sequence database into a smaller pool of template HMM profiles for HMM-HMM alignment using the Viterbi algorithm. (Source: Alchemy Bio)
Instead of representing every column in the query HMM as a vector of 20 probabilities corresponding to each amino acid, HHblits converts this vector into a single character from an alphabet of 219 letters (corresponding to all ASCII characters). Each letter represents the probability distribution of a typical column in a profile HMM, reducing the infinite number of probability distributions to only 219 representative distributions.
Alphabet of all 219 representative probability distributions of columns in a profile HMM. (Source)
To compute the letter with the distribution closest to the true distribution of column i in the query HMM, we compute the following score:
where q_i is the emission probability at column i of the query HMM and p_k is the probability from the kth letter in the alphabet.
Using the extended query sequence, RF leverages HHblits to iteratively construct an N x L multiple sequence alignment and then runs HHsearch to find a set of template structures with the following steps:
The extended query profileis compared to a database of template profile sequences (where each column of the template profiles is converted into one of 219 representative distributions) and the log-odds score of the longest consecutive (gapless) alignment is computed. The scores above 2.5 + lg(N_qN_t), where N_q is the length of the query sequence and N_tis the length of the target sequence, are passed to the second prefilter.
The second prefilter computes a score for the gapped alignment between the query extended sequence and target HMM using the Smith-Waterman alignment which scores the first gap transition (called gap open) with a higher penalty (5 bits) and subsequent transitions in the same gap (gap extension) with a smaller penalty (1 bit). This bit score is converted into an E-value with the following equation:
\(E=N_{db}L_qL_t\times \underbrace{2^{-S}}_{S\text{: bit score}}\)
where N_db is the number of sequences per HMM in the database. Only sequences with E-values below 1000 are passed to the next step. The two-stage prefilter results in a 10- to 100-fold reduction in the pool of sequences.
Then, we run the Viterbi algorithm on the reduced pool of template HMMs and add the template sequences with the highest LSO scores to the MSA. We iteratively repeat this process, updating the MSA with new sequences until the MSA contains 2000 sequences with at least 75% sequence coverage or 5000 sequences with at least 50% sequence coverage.
Finally, the output query MSA from HHblits is passed into the HHsearch algorithm that performs HMM-HMM alignment with template HMMs of sequences with known structure from the PDB100 database. HHsearch refines the query MSA and outputs the top 10 template sequences and their structures which is used as input to the pair-wise feature module.
MSA Embeddings
The N x L MSA alignment matrix is transformed into an N x L x d_msa array of feature embeddings from a set of 20 learned embeddings for each residue type. Each embedding is added to a 1-dimensional positional encoding indicating the position of the residue along its sequence as well as a query encoding that distinguishes residues in the query sequence and those in the template sequences.
1-Dimensional Positional Encoding
When computing attention, the model considers the feature embeddings as an unordered set of residue features and has no inherent knowledge of the position of the residue in the sequence. Thus, positional encodings are added to feature arrays to inform the model of the position of the residue that attention is being calculated for and the relative distance of the residues that it is attending to.
The 1-dimensional positional encoding is a d-dimensional vector, where d is the dimension of the feature embedding. For the residue at position i in the protein, the positional encoding (p_i) is defined as:
For d = 64, the function for ωₖ for values of k ∈ [0, d/2] can be visualized graphically as:
Graph of the change in frequency ωₖ as k increases. (Source: Alchemy Bio)
When the frequency ωₖ is equal to 1, the period of the sinusoidal function 2π, which is the minimum period. As the value of ωₖ decreases exponentially and approaches 0, the frequency decreases and the period increases, which can be observed as the smoother changes in color down a column of the matrix below.
Positional embedding matrix for a sequence with length 50 (L = 50) and the embedding dimension of 128 (d = 128), where every row represents the positional encoding for the residue at position i. The values are between -1 (red) and 1 (blue) and 0 is white. (Source)
Therefore, the positional encoding is a vector of pairs of sine and cosine of decreasing frequency along the sequence dimension evaluated at position i.
You may be wondering:why can’t we simply add the index i of the position to each entry of the embedding?
For longer sequences, the index i can take extremely large integer values. Adding large integers to the position embeddings can dilute the signal of the original embedding that the model should be learning from. This also expands the scale of the embeddings to a large range of values which makes it difficult for the model to learn patterns across the embeddings and reach convergence.
Since the values of sine and cosine are bounded between [0, 1], adding a positional encoding with sine and cosine entries maintains the original scale of the embeddings while effectively encoding absolute and relative positional information.
Another reason for defining the positional encoding with sinusoidal functions is because we can extract the positional encoding for a position i + Δ that is a fixed distance Δ away from any position i by multiplying the sine/cosine pair for each value of k by a linear transformation matrix M dependent only on k and Δ (not dependent on i).
This provides valuable context into the relative distances between two positions for the model to learn the separations between residues with generally stronger or weaker interactions across training examples and use it to compute attention scores with higher accuracy.
An intuitive interpretation of the positional encoding is to consider every pair of sine/cosine entries as defining the angle of a single hand on a clock (hour hand, minute hand, second hand, etc.). The positional encoding has a total of d/2 hands, which each rotate at a distinct frequency of ωₖ.
Moving from position i along the residue dimension by Δ is like waiting some time Δ. The positional encoding for position i + Δ along the sequence is the sine/cosine components for all the clock’s hands at their new positions after rotating at different frequencies.
Since the frequency of each hand is constant, it always rotates by a constant angle in a defined increment of time Δ. This explains why we can derive the kth sine and cosine entries of the positional encoding of a position i + Δ simply by multiplying each sine/cosine pair of the encoding for position i by a rotation matrixM that essentially rotates the angle of each hand by a fixed angle defined only by the time increment Δ and its frequency ωₖ. (analogy from a comment on this blog article)
We define the PositionalEncoding module that adds a positional encoding to every embedding in an input feature array x with shape (B, L, d). The constructor generates the positional encodings up to a maximum sequence length of 5000 with the following code:
pe is an array with shape (max_len, d_model) initialized to store the precomputed d-dimensional positional encodings for sequences up to a length of max_len.
position is an array with shape (max_len, 1) with the indices i = [0, max_len-1] to be multiplied to the array of ω values and inputted into the sine and cosine functions.
div_term is a d/2-dimensional vector that stores the ω values computed for k ranging from 1 to d/2 which are decreasing exponentially. This vector is multiplied by the position array to generate the inputs to the sine and cosine functions.
torch.arange(0, d_model, 2) generates a (d/2)-dimensional vector with all even numbers ranging from 0 to d_model. This corresponds to the 2k term in the numerator of the equation. It is then multiplied by ln(10000) and divided by the constant d. The negative sign ensures that the function is exponentially decreasing and is bounded between 0 and 1.
pe[:, 0::2] = torch.sin(position * div_term) sets all even indices of the positional encoding to the sin(ωₖ*i) for all values of k. pe[:, 0::2] aligns the lth index of the (d/2)-dimensional vector to the 2lth index of the pe vector.
pe[:, 1::2] = torch.cos(position * div_term) sets all the odd indices of the positional encoding to the cos(ωₖ*i) for all values of k.
pe = pe.unsqueeze(0) adds inserts a singleton dimension to convert the shape to (1, max_len, d_model) which will be broadcasted to match the batch dimension when added to the input feature array.
self.register_buffer('pe', pe) sets pe to a buffer, which is part of the module’s state that is not updated during gradient descent.
We then define the forward function that takes as input the feature array x and an array containing the indices of the positions along the sequence dimension of the feature array that require the addition of a positional encoding idx_s. This reduces the positional encoding matrix from (B, max_len, d) to (B, L, d) so it can be added easily to the input feature array of the same shape.
We will initialize and call the PositionalEncoding class for all arrays of single residue features (notably MSA embeddings) that are fed into an attention block. Later, we will see how this idea extends to 2D positional encodings.
Query Encoding
To distinguish the MSA embeddings of the query sequence from the template sequences when computing attention, we add a learned query encoding to each MSA embedding along the query sequence and a learned template encoding to each embedding along all template sequences.
Adding a learned query encoding to each MSA embedding in the query sequence and a learned template encoding to each position in each template sequence. (Source: Alchemy Bio)
To do this, we define a QueryEncoding module that initializes a lookup dictionary of two d_model-dimensional encodings of learnable weights using nn.Embedding(2, d_model), one that will be added to the query embeddings and another that will be added to each of the template embeddings in the MSA. The learnable entries of the query and template encodings will be optimized during gradient descent.
The forward function takes the MSA array with shape (B, N, L, d_msa) and adds the query/template encodings.
First, we initialize an array of 1s with shape (B, N, L) that will store the indices of the encoding (either 0 for query or 1 for template) that will be added to the MSA embedding of each residue in the MSA. We set the entries at index 0 along the sequence dimension to 0 since they correspond to query embeddings.
Now, we can generate an array of query/template encodings with shape (B, N, L, d_msa) by calling self.pe(idx) which returns an array with the d_msa-dimensional query encoding for entries of 0 in idx or the template encoding for entries of 1 in idx. We add the encoding array to the array of MSA embeddings.
Initializing the MSA Embeddings
After generating the array containing the indices of each amino acid in the MSA with shape (B, N, L), we feed it into the MSA_emb module that returns a d_msa-dimensional learned embedding corresponding to the type of amino acid at each position.
We create an nn.Embedding dictionary that stores 21 d_msa- dimensional embedding vectors corresponding to each of the 21 common amino acids. Each amino acid embedding is learned during model training.
Then, we add the 1D positional encoding to each embedding, a learned query encoding to each position in the target sequence, and a learned template encoding to each position in the template sequences to get an initial MSA embedding array.
Now we will see how to incorporate contextual features across the MSA into each embedding using axial attention.
Axial Attention
Given a 2-dimensional array as input, axial attention reduces computational time and space by restricting the receptive field that each embedding of the input array attends to down to either the row (row-wise attention) or the column (column-wise attention) that it is in. A combination of row-wise and column-wise axial attention expands the receptive field such that each embedding of the array to attend to every other embedding in the array directly or indirectly.
If you would like an introduction to the attention mechanism, I encourage you to read my previous article, where I break down attention on amino acid sequences and SMILES strings for drug-target interaction prediction.
Here, I will be skipping over the basic ideas of attention, including the intuition behind query, key, and value projections and the rationale behind multi-head attention. Instead, I will focus on multiple variations of the original attention mechanism, and how they are used to process and pass information across multi-dimensional data.
Multi-Head Axial Attention
To apply regular multi-head attention on the MSA embeddings, every embedding in the N x Lx d_msa matrix would attend to every other embedding, equivalent to executing attention on a flattened (N*L)-lengthsequence.
With Big-O notation2, the time and space complexities of regular multi-head attention are bounded by:
To derive the time complexity, we observe that every entry of the (N*L) x (N*L) matrix of attention scores is computed by taking the dot product between the d_msa-dimensional query and key vectors. Since the dot product operation requires computing d_msa along the elements of the vectors, this results in a total of (N*L)²d_msa computations.
The space complexity is derived from storing the three(N*L) x d_msa matrices of query, key, and value vectors and the (N*L) x (N*L) matrix of attention scores.
This results in a time and space complexity that grows quadratically as the length of the protein sequence (L) or the number of templates in the MSA (N) increases. For stochastic gradient descent, this will increase even faster with large batch sizes (B).
By restricting the range of elements that each element attends to only a single row or column, the computation and memory required decreases dramatically.
For row-wise axial attention along the residue dimension of width L, the time and space complexities are reduced to:
Column-wise axial attention and the time complexities of each step. (Source: Alchemy Bio)
Applying row-wise attention followed by column-wise attention or vice versa allows every residue in the MSA to directly attend to every other residue within their sequenceand column while indirectly attending to every other residue outside their sequence or column.
For the rest of this section, we will refer to row-wise attention as residue-wise attention and column-wise attention as sequence-wise attention.
In the diagram below, a single embedding at sequence n and position i in the MSA first attends to all other positions in the same sequence n. Likewise, the position i in every other sequence also attends to the embeddings within their sequence (row-wise attention). Then, the position i at sequence n attends to every other embedding in column i across all N sequences in the MSA that have already attended to every position within their own sequence.
Axial attention with residue-wise attention across each row and sequence-wise attention across each column allows each embedding to directly or implicitly attend to every other residue in the N x L MSA. (Source: Alchemy Bio)
In addition, axial attention automatically places greater weight on residues within the same sequence or column whereas regular attention across the entire matrix lacks the baseline context that would enable the model to distinguish between a residue in the same sequence to one in a different sequence.
Residue-wise multi-head attention for position i in sequence n can be summarized as projecting the MSA embedding for position i into a query (q) vector and all L positions into key (k), and value (v) d-dimensional vectors and splitting each vector into Hd/H-dimensional equal-sized subvectors for multi-head attention (we denote d_h = d/H as the embedding dimension at each head).
\(\begin{align}\mathbf{q}_{n,i}&=\mathbf{W}^{Q}\mathbf{f}^{\text{msa}}_{n,i}+\mathbf{b}^{Q}_{n}\tag{$\mathbf{q}\in\mathbb{R}^{d_{\text{msa}}}$}\\\{\mathbf{k}_{n,j}\}_{j=1}^L&=\mathbf{W}^{K}\mathbf{f}^{\text{msa}}_{n,j}+\mathbf{b}^{K}_{n}\tag{$\mathbf{W}^K\in\mathbb{R}^{d_{\text{msa}}\times d_{\text{msa}}}$}\\\{\mathbf{v}_{n,j}\}_{j=1}^L&=\mathbf{W}^{V}\mathbf{f}^{\text{msa}}_{n,j}+\mathbf{b}^{V}_{n}\tag{$\mathbf{W}^V\in\mathbb{R}^{d_{\text{msa}}\times d_{\text{msa}}}$}\\\nonumber\\\mathbf{q}_{n,i},\mathbf{k}_{n,j},\mathbf{v}_{n,j}&\xrightarrow{\text{split to }H\text{ heads}}\{\mathbf{q}^h_{n,i},\mathbf{k}^h_{n,j},\mathbf{v}^h_{n,j}\}_{h=1}^H\tag{$d_h=d/H$}\\\nonumber\end{align}\)
Then, we compute a set of L attention weights for each attention head h by taking the dot product of the query vector with each key vector and scaling down by the square root of d_h. The updated MSA embedding at each head is a weighted average of the value vectors for each of the positions in the sequence that we concatenate into a single d-dimensional vector and process with a final linear transformation layer to get the updated MSA embedding.
Multi-head residue-wise attention for position i in sequence n attending to all other positions j in the same sequence in a total of H representation subspaces. (Source: Alchemy Bio)
Column-wise attention has the same steps as row-wise attention, except instead of attending to every position along the sequence, the residue at position i attends to the other residues at position i across all N sequences in the MSA (indexed by m).
\(\begin{align}\mathbf{q}_{n,i}&=\mathbf{W}^{Q}\mathbf{f}^{\text{msa}}_{n,i}+\mathbf{b}^{Q}\tag{$\mathbf{q}\in\mathbb{R}^{d_{\text{msa}}}$}\\\{\mathbf{k}_{m,i}\}_{m=1}^N&=\mathbf{W}^{K}\mathbf{f}^{\text{msa}}_{m,i}+\mathbf{b}^{K}\tag{$\mathbf{W}^K\in\mathbb{R}^{d_{\text{msa}}\times d_{\text{msa}}}$}\\\{\mathbf{v}_{m,i}\}_{m=1}^N&=\mathbf{W}^{V}\mathbf{f}^{\text{msa}}_{m,i}+\mathbf{b}^{V}\tag{$\mathbf{W}^V\in\mathbb{R}^{d_{\text{msa}}\times d_{\text{msa}}}$}\\\nonumber\\\mathbf{q}_{n,i},\mathbf{k}_{m,i},\mathbf{v}_{m,i}&\xrightarrow{\text{split to }H\text{ heads}}\{\mathbf{q}^h_{n,i},\mathbf{k}^h_{m,i},\mathbf{v}^h_{m,i}\}_{h=1}^H\tag{$d_h=d/H$}\\\nonumber\end{align}\)
Let’s define the MultiheadAttention module that can be used for both row- and column-wise attention simply by permuting the shape of the input feature array.
In the constructor, we initialize the dimension of the embeddings at each attention head, the WQ, WK, and WV linear transformation layers that project each MSA embedding to q, k, and v vectors, and the final output linear transformation layer WO that processes the concatenated embeddings from each attention head.
Later, when we discuss cross-attention, the feature array used to generate the query embedddings is not equal to the feature array used to generate the key and value embeddings. This is why the module below defines additional variables k_dim and v_dim that store the dimension of the embeddings used to generate the key and value vectors seperately. In this section, we assume that k_dim = v_dim = d_model= d_msa.
The forward function takes the feature array used to generate the query, key, and value embeddings reshaped such that the last two dimensions are (axial_dim, d_model) and all other dimensions are concatenated to the batch dimension so attention for every row or column can be computed in parallel.
For row-wise (residue-wise) attention, the column dimension is concatenated with the batch dimension to shape (B*N, L, d_msa) to compute attention across each sequence in the MSA for all query sequences in the training batch.
For column-wise (sequence-wise) attention, the row dimension is concatenated with the batch dimension to shape (B*L, N, d_msa) to compute attention across each column in the MSA for all query sequences in the batch.
Now, we will implement the forward function in the context of column-wise attention, where the input is the initial (B*L, N, d_msa) array of MSA embeddings:
First, we compute the q, k, and v vectors for all embeddings in the MSA in parallel by feeding the (B*L, N, d_msa) array through three distinct linear transformation layers that transform each MSA embedding with a distinct d_msa x d_msa set of learnable weights.
Then, we split each q, k, and v embedding into Hd_h-dimensional vectors, by reshaping the Q, K, and V arrays to shape (B*L, N, H, d_h) and then permuting the dimensions to (B*L, H, N, d_h) so that the last two dimensions are an N x d_h matrix of q, k, or v vectors for a single attention head.
\(\underbrace{\begin{bmatrix}—&\mathbf{q}_{1,i}&—\\—&\mathbf{q}_{2,i}&—\\&\vdots\\—&\mathbf{q}_{N,i}&—\end{bmatrix}}_{\mathbf{Q}_i}\xrightarrow{\text{split into }H\text{ heads}}\left\{\underbrace{\begin{bmatrix}—&\mathbf{q}^h_{1,i}&—\\—&\mathbf{q}^h_{2,i}&—\\&\vdots\\—&\mathbf{q}^h_{N,i}&—\end{bmatrix}}_{\mathbf{Q}^h_i}\right\}_{h=1}^H\)
The code below generalizes for axial attention across any axis and for different source (key and value vectors) and target (query vectors) feature embeddings with different dimensions. For column-wise attention for MSA to MSA updates, both the source and target features are MSA embeddings so tgt_axial_dim = src_axial_dim = N.
At each attention head, the set of attention scores for the residue at position i is computed in parallel by multiplying the N x d_h query matrix Q with the transposed d_h x N key matrix K. This produces an N x N matrix of raw attention scores at the attention head h.
In the code implementation, multiplying the query array with shape (B*L, H, N, d_h) with the key array transposed along the last two dimensions (B*L, H, d_h, N) results in an array of attention scores with shape (B*L, H, N, N).
Since each row of the attention matrix is the set of N attention scores to compute the contextual embedding for position i attending to every other sequence in the MSA, we apply the softmax function such that each row of attention scores adds up to 1.
Each row of the N x N attention matrix is used to compute the weighted average of the set of N value vectors for position i that becomes the contextual embedding for a single sequence n at head h. We parallelize this process to compute the contextual embeddings position i for n = [1…N] by taking the matrix product of the N x N attention matrix and N x d_h value matrix V.
This is the same as multiplying the (B*L, H, N, N) attention array with the (B*L, H, N, d_h) value array, to get an array with shape (B*L, H, N, d_h), where each head has a corresponding N x d_h matrix of contextual embeddings for position i.
To combine the contextual embeddings across all H heads, we reshape the output array to (B*L, N, H, d_h), separating the embeddings of each residue, and concatenate all H embeddings for each residue by squeezing the last two dimensions to get an output array with shape (B*L, N, d_msa), where d_msa = H*d_h.
Finally, we process the d_msa-dimensional vector concatenation of the contextual embeddings generated from each attention head with a d_msa x d_msa linear transformation layer.
If you consider each attention head as learning a single representation subspace describing the relationship between homologously aligned residues, the linear layer learns the relative importance of each representation subspace to generate the final contextual embedding.
Now, let’s see how we can take full advantage of the homologous properties of the MSA to incorporate additional contextual information into the attention mechanism using tied axial attention.
Tied Axial Attention
Since homologs have similar pair-wise interactions between aligned positions along the residue dimension, tied residue-wise attention computes a single set of attention scores for each column of the MSA that is shared across all sequences to compute residue-wise axial attention.
Residue-wise attention calculates attention scores for each ordered pair of residue positions within each sequence in the MSA, represented by a row in the N x L MSA matrix.
Since the MSA module aligns homologs such that each column is the best alignment between the template sequence with the query sequence, the chemical relationship between the residue along column i of the MSA with the residue at another column j should be similar in each sequence in the MSA.
Consider a pair of homologous proteins A and B, where the residue at position 1 of protein A has a strong interaction with the residue at position 4. Since A and B are homologs with shared structural motifs, it is likely that the residues at position 1 and position 4 of protein B also have a strong interaction.
Tied attention introduced in the MSA Transformer by Rao et al. leverages the similarity between homologous sequences by computing a shared set of attention scores to generate the contextual embedding for every amino acid at a given position along the residue dimension across all sequences in the MSA. In other words, the attention scores remain constant for each sequence in the MSA.
\(\underbrace{\mathbf{q}_{n,i}^h\cdot\mathbf{k}_{n,j}^h}_{\text{distinct for each sequence }n}\xrightarrow{\text{tied residue-wise attention}}\underbrace{\sum_{n=1}^N\mathbf{q}_{n,i}^h\cdot\mathbf{k}_{n,j}^h}_{\text{shared across all }N\text{ sequences}}\)
Tied residue-wise attention at a single attention head h for a single position i attending to all other positions j within their sequence. The attention scores are computed at each of the N sequences and summed together. Then, the aggregated attention scores are used to scale the value vectors at each sequence separately, producing the updated MSA embeddings. (Source: Alchemy Bio)
For a position i attending to position j along the residue dimension of the nth sequence of the MSA, the untied attention score for a single head in multi-head attention is calculated by taking the dot product between the query embedding at position i with the key embedding at position j.
\(\underbrace{\mathbf{q}^h_{n,i}\cdot \mathbf{k}^h_{n,j}}_{\text{untied attention score for position }i\text{ attending to position }j}\)
The tied attention score is the sum of the attention scores for the positions i attending to j for all N sequences in the MSA.
\(\underbrace{\sum _{n=1}^N\mathbf{q}^h_{n,i}\cdot \mathbf{k}^h_{n,j}}_{\text{tied attention score for position }i\text{ attending to position }j}\)
Since each score is computed as the sum of N dot products, which itself is the sum of d_h element-wise products, we scale down each tied attention score by √N*d_hto normalize the score to its original scale.
Finally, we apply the softmax function across the set of L tied attention scores for each position attending to all other positions along the residue dimension.
\(\alpha_{ij}^h=\underbrace{\frac{\exp(\frac{\sum _{n=1}^N\mathbf{q}^h_{n,i}\cdot \mathbf{k}^h_{n,j}}{\sqrt{Nd_h}})}{\sum_{j'=1}^{L}\exp(\frac{\sum _{n=1}^N\mathbf{q}^h_{n,i}\cdot \mathbf{k}^h_{n,j'}}{\sqrt{Nd_h}})}}_{\text{softmax across scores for position }i}\)
To get the output embedding for position i in a sequence n, we take the weighted sum of the input MSA embeddings for all L positions in sequence n scaled by the set of L tied attention scores. Like multi-head axial attention, the output contextual embeddings across all H heads are concatenated and processed with a linear layer.
Even though each contextual embedding still only attends to the positions within their sequence, the tied attention scores share information on how strongly each position should attend to every other position across homologs.
Below is the visualization for the matrix implementation of tied attention, which we will translate into code.
Tied residue-wise attention with matrix multiplication. After computing the attention scores at each sequence, they are summed together to get a single L x L matrix of tied attention scores. The tied attention scores are multiplied with the L x d_msa value matrix at each sequence to get the updated MSA embeddings. (Source: Alchemy Bio)
The implementation of the TiedMultiheadAttention module is similar to that of MultiheadAttention with a few key differences:
Instead of concatenating the non-axial dimension to the batch dimension, we leave the MSA embedding array with shape (B, N, L, d_msa) when projecting to q, k, and v vectors so that we can sum across the sequence dimension N when computing the tied attention weights.
We split the vectors into H d_h-dimensional subvectors for each head by reshaping the array to (B, N, L, H, d_h) and permute to shape (B, N, H, L, d_h), so we have a L x d_h matrix of q, k, v vectors at each head for each sequence in the MSA.
We multiply the query array with the key array transposed along the last two dimensions to get an array with shape (B, N, H, L, L) containing a total of H L x L attention matrices for each sequence in the MSA.
To get the tied attention scores for each attention head, we take the element-wise sum across the sequence dimension N of the attention array, converting from a (B, N, H, L, L) array to a (B, H, L, L) array. At each head, there is a single L x L matrix of tied attention scores, each of which captures a representation subspace shared across homologs.
Alternatively, Steps 3 and 4 can be combined with torch.einsum:
To get the final attention scores, we apply softmax along the last dimension of the array of tied attention scores so that every row of the L x L matrix at each head adds up to 1.
At each attention head, we multiply the L x d_h matrix of value embeddings for every sequence in the MSA with the same L x L matrix of tied attention scores.
where each contextual embedding for position i in sequence n at head h is the linear combination of the set of value vectors for sequence n scaled by the tied attention weight for i attending to each position j.
\(\mathbf{f}^h_{n,i}=\underbrace{\sum_{j=1}^L\underbrace{\text{SM}_i\left(\sum_{n=1}^N\mathbf{q}^h_{n,i}\cdot \mathbf{k}^h_{n,j}\right)}_{\text{attention score}}\underbrace{\mathbf{v}^h_{n,j}}_{\text{value vector}}}_{\text{weighted average of value vectors}}\)
To implement this, we must add a singleton dimension to convert the attention array from shape (B, H, L, L) to (B, 1, H, L, L), where the second dimension will be broadcasted to match the dimension of N. The (H, L, L) array of tied attention weights is repeated N times along the sequence dimension to shape (B, N, H, L, L) before being multiplied with the value array of shape (B, N, H, L, d_h) to get the (B, N, H, L, d_h) array of contextual embeddings.
To concatenate all H contextual embeddings for each residue in the MSA, we permute the dimensions of the output array so that the last two dimensions correspond to the H x d_h matrix of contextual embeddings for each residue and flatten the last two dimensions.
In addition to the updated MSA embeddings, the TiedMultiheadAttention module also returns the L x L x H matrix of tied attention scores for each pair of residues ij that will be used to update the pair features.
Since attention scores are not symmetrical (ie. the score for i attending to j is not equal to the score for j attending to i), we add the original L x L attention matrix with the transposed attention matrix and multiply each element by 0.5 to scale the sum back to its original scale.
Then, we reshape the attention array to shape (B, L, L, H) so that for every pair of positions i and j, there is a corresponding H-dimensional vector of the symmetrical attention scores from every attention head.
\(\vec{\alpha}_{ij}=\vec{\alpha}_{ji}=\underbrace{\begin{bmatrix}\frac{1}{2}\left(\alpha^1_{ij}+\alpha^1_{ji}\right)\\\\\frac{1}{2}\left(\alpha^2_{ij}+\alpha^2_{ji}\right)\\\vdots\\\frac{1}{2}\left(\alpha^H_{ij}+\alpha^H_{ji}\right)\end{bmatrix}}_{\text{pair features for positions }ij}\in \mathbb{R}^H\)
Finally, the TiedMultiheadAttention module returns the updated MSA embedding array with shape (B, N, L, d_msa) and the pair attention features with shape (B, L, L, H).
We can also implement tiedsequence-wise attention along each column of the MSA, where the attention scores are shared across all positions in a single sequence ninstead of a single position. This means that the set of N attention scores used to attend sequence n to every other sequence m in column i is equal to the set of attention scores used to attend sequence n to every other sequence m in column j.
\(\underbrace{\mathbf{q}_{n,i}^h\cdot\mathbf{k}_{m,i}^h}_{\text{distinct for each column }i}\xrightarrow{\text{tied sequence-wise attention}}\underbrace{\sum_{i=1}^L\mathbf{q}_{n,i}^h\cdot\mathbf{k}_{m,i}^h}_{\text{shared across all }L\text{ columns}}\)
Tied sequence-wise attention at a single attention head h for a single sequence n attending to all other sequences m at position i. The attention scores are computed at each of the L columns and summed together. Then, the aggregated attention scores are used to scale the value vectors at each position separately, producing the updated MSA embeddings. (Source: Alchemy Bio)
Since the set of N tied attention scores is distinct for each sequencein the MSA, this ensures that each residue in a a sequence n attends to each sequence in the MSA the same way based on the sequence-wide relationship of sequence n with every other sequence m.
The implementation for tied sequence-wise attention uses the same TiedMultiheadAttention module but we permute the dimensions of the input array to (B, L, H, N, d_h) so attention is computed across each column and the attention scores are summed over the residue dimension L instead of the sequence dimension.
Soft-Tied Residue-Wise Attention
Building off of tied attention, RF introduces soft-tied attention, where instead of taking a simple sum over the attention scores from all N sequences in the MSA, the attention scores generated at each sequence are weighted based on the strength of alignment with the query sequence, and each soft-tied attention score is the weighted average across all sequences.
Since not all template sequences are strongly aligned to every position in the query sequence, computing the attention scores for each position as a simple sum across all sequences can result in unaligned sequences contributing attention scores that contain irrelevant information on the relationship with other positions in the sequence.
Thus, the RF model introduces soft-tied multi-head attention by computing theshared attention scores as a weighted average of the attention scores across each template sequence. At a given position, templates with strong co-evolution with the query sequence are weighted more heavily than templates with weak co-evolution based on a set of sequence weights.
In the experiments for RF, the L x L matrices of soft-tied attention scores for each pair of residues (red boxes in Panel B) resembled the true contact maps of proteins (Panel A). These contact maps are masked to show only residue pairs within a specified distance in the 3D protein structure, demonstrating the ability of 1D features like MSA embeddings to learn complex 3D interactions between residues.
In addition, some attention maps showed stronger attention to only residues without close contacts (blue boxes), indicating that the model can distinguish features of non-interacting residue pairs.
The L x L residue pair-wise attention maps generated from soft-tied residue-wise attention resemble the true contact map of the CASP14 target protein T1049 (panel A). Yellow represents residues within a specified distance in the 3D protein structure. The attention maps in red boxes resemble true contact maps and the ones in blue boxes only attend to residues without true contacts. (Source: Supplementary Figure S12)
Soft-tied attention applies a set of Nsequence weights that reflect the degree of alignment of a given position i across all N sequences in the MSA. For a single position i in the MSA, instead of generating a set of L tied attention scores that attend to every other position by adding the attention scores across all N sequences, we generate the “soft-tied” attention score that attends to another position j by taking the weighted sum of the ij attention scores across all N sequences scaled by the set of N sequence weights for the position i. To emphasize, we use the set of N sequence weights for the position that we are calculating attention for, not the position that is being attended to.
Before we break down the implementation for soft-tied attention, we need to take a slight detour to discuss how the sequence weights are generated.
The SequenceWeights module calculates an N x L matrix of sequence weights, where the nth row corresponds to the set of L weights that will scale the set of attention scores generated for each of the L positions in the nth sequence in the MSA.
These sequence weights are generated similarly to attention scores by transforming the MSA embeddings for each position in the target sequence into L query embeddings transforming the embeddings of each position in every homologous sequence into N x L key embeddings and taking the dot product of query embedding (query sequence) with all N key embeddings for the position i. This generates a similarity score that measures the conservation or co-evolution of the residue at a position i in the target sequence with the residues at the same position i in each of the homologous sequences.
The set of N sequence weights for a single position i in the query sequence is computed by projecting the MSA embedding for residue i in the target (or query) sequence into a query vector and the embedding for residue i of the template sequences into key vectors and computing the scaled dot product attention scores. The sequence weights are used to compute a single set of soft-tied attention scores, where each score is a weighted average of the attention scores across all N sequences scaled by their sequence weight. (Source: Alchemy Bio)
To distinguish the query and key embeddings used to generate the sequence weights from those used to generate the attention scores, we will use a superscript w to denote the query and key embeddings for sequence weights.
We also denote the MSA embeddings for each position in the target (or query) sequence with the subscript 1 since it is the first sequence in the MSA. We index the embeddings for all remaining template sequences in the MSA that will be transformed into key vectors with the subscripts ranging from 2 to N.
Since sequence weights are computed separately for each column of the MSA alignment or each position in the query sequence, we permute the dimensions of the MSA feature array to shape (B, L, N, d_msa).
Next, we extract the MSA embeddings for all L positions of the targetsequence by extracting the first element along the sequence dimensionto get an array with shape (B, L, d_msa). We then insert a singleton dimension to get an array with shape (B, L, 1, d_msa), where the query vector for each position is a row vector for matrix multiplication.
After extracting the (B, L, 1, d_msa) array of MSA embeddings of every position in the target sequence, we project all L embeddings into a d_msa-dimensional query vector with a linear transformation layer and then split each vector into H equal sub-vectors for each attention head.
For column i in the MSA, we have d_h-dimensional query vector at each head that we will be used to compute the dot product sequence weights that indicate the strength of alignment of the query sequence at position i and each of the template sequences (including itself). If position i of the query sequence is has a strong alignment with the nth template sequenceof the MSA, the (i, n)th sequence weight will be large.
\(\begin{align}\mathbf{q}^w_{1,i}&=\mathbf{f}^{\text{msa}}_{1,i}\mathbf{W}^Q+\mathbf{b}^Q\tag{$\mathbf{W}^Q\in\mathbb{R}^{d_{\text{msa}}\times d_{\text{msa}}}$}\\\mathbf{q}^w_{1,i}&\xrightarrow{\text{split into }H\text{ heads}}\{\mathbf{q}^{w,h}_{1,i}\}_{h=1}^H\end{align}\)
Then, we project the full MSA embedding array with shape (B, N, L, d_msa) to an array of key embeddings with shape (B, N, L, d_msa) which we split into H heads to get an array with shape (B, N, L, H, d_h).
For a each position i, we compute a N x d_h matrix of key vectors at each head that will be used to compute the strength of alignment of position i of the query sequence with each template sequence in the MSA.
\(\begin{align}\underbrace{\begin{bmatrix}—&\mathbf{f}^{\text{msa}}_{1,i}&—\\—&\mathbf{f}^{\text{msa}}_{2,i}&—\\&\vdots\\—&\mathbf{f}^{\text{msa}}_{N,i}&—\end{bmatrix}}_{\text{MSA embeddings for all sequences}}
\mathbf{W}^K+\mathbf{b}^K&=\underbrace{\begin{bmatrix}—&\mathbf{k}^{w}_{1,i}&—\\—&\mathbf{k}^w_{2,i}&—\\&\vdots\\—&\mathbf{k}^w_{N,i}&—\end{bmatrix}}_{\text{sequence weight keys }\mathbf{K}^w_i}\\\begin{bmatrix}—&\mathbf{k}^{w}_{1,i}&—\\—&\mathbf{k}^w_{2,i}&—\\&\vdots\\—&\mathbf{k}^w_{N,i}&—\end{bmatrix}\xrightarrow{\text{split into }H\text{ heads}}&\left\{\begin{bmatrix}—&\mathbf{k}^{w,h}_{1,i}&—\\—&\mathbf{k}^{w,h}_{2,i}&—\\&\vdots\\—&\mathbf{k}^{w,h}_{N,i}&—\end{bmatrix}\right\}_{h=1}^H\end{align}
\)
We then take the matrix product of the 1 x d_h query row-vector and transpose d_h x N key matrix to get an N-dimensional row vector of raw sequence weights for each position i for each attention head. This is equivalent to computing the dot product between the query vector for position i from the target sequence with the set of N key vectors for position i from each sequence in the MSA.
\(\begin{bmatrix}—&\mathbf{q}^{w,h}_{1,i}&—\end{bmatrix}\underbrace{\begin{bmatrix}|&|&&|\\\mathbf{k}^{w,h}_{1,i}&\mathbf{k}^{w,h}_{2,i}&\dots&\mathbf{k}^{w,h}_{N,i} \\|&|&&|\\\end{bmatrix}}_{\mathbf{K}^{w,h\top}_i}=\underbrace{\begin{bmatrix}\mathbf{q}^{w,h}_{1,i}\cdot \mathbf{k}^{w,h}_{1,i}&\mathbf{q}^{w,h}_{1,i}\cdot \mathbf{k}^{w,h}_{2,i}&\dots &\mathbf{q}^{w,h}_{1,i}\cdot \mathbf{k}^{w,h}_{N,i}\end{bmatrix}}_{N\text{-dimensional vector of raw sequence weights for position }i}\)
In the implementation, we align the dimensions for matrix multiplication by reshaping the query array to (B, L, H, 1, d_h) and the key array to (B, L, H, d_h, N) and compute the array product using torch.matmul to get an array with shape (B, L, H, 1, N) of raw sequence weights.
Now, we normalize each weight by √d_h and apply softmax separately to each set of N sequence weights for each position and each head.
The output of the SequenceWeights module is an array with shape (B, L, H, 1, N) array of sequence weights with dropout.
Now that we know how the set of N sequence weights for each position i along the query sequence is computed, we can use them for soft-tied multi-head attention.
Soft-tied residue-wise attention block for attention on position i. First, we compute the set of N sequence weights for position i. Then, we compute the set of L attention scores at each sequence and take the weighted average. Finally, the shared set of soft-tied attention scores are used to scale the value vectors at each sequence to generate the updated MSA embedding. (Source: Alchemy Bio)
Unlike tied attention scores which are computed by taking the sum of theattention scores for position i attending to position j across all N sequences in the MSA, soft-tied attention scores are the weighted sum of the attention scores for position i attending to position j for each of sequence in the MSA scaled by the (n, i)th sequence weight.
\(\underbrace{\sum _{n=1}^N\mathbf{q}^h_{n,i}\cdot \mathbf{k}^h_{n,j}}_{\text{tied attention score for }i\text{ attending to }j}\xrightarrow{\text{scale with sequence weights}}\underbrace{\sum _{n=1}^Nw^h_{n,i}\left(\mathbf{q}^h_{n,i}\cdot \mathbf{k}^h_{n,j}\right)}_{\text{soft-tied attention score for }i\text{ attending to }j}\)
Since the (n, i)th sequence weight is a measure of how strongly the alignment is for position i between the target and nth template sequence, it is used to determine how much the attention scores for position i of the nth sequence contribute to the soft-tied attention scores for position i that will be shared when computing row-wise attention.
The sequence weights are normalized such that the set of N sequence weights for a single position i add up to 1, so we only have to scale each attention score down by √d_h instead of √N*d_h (tied attention).
The normalized soft-tied attention score for position i attending to position j that is shared when computing residue-wise attention for all N sequences at the attention head h is given by the equation below:
Let’s break down the implementation of the SoftTiedMultiheadAttention module that uses the SequenceWeights class to compute the soft-tied attention scores which are used to perform attention on the MSA feature array with shape (B, N, L, d_msa).
First, we generate a (B, L, H, 1, N) array of sequence weights by calling the instance of the SequenceWeights module defined in the constructor with the MSA feature array as input. Then, we permute the dimensions of the array to shape (B, N, H, L, 1) so that the singleton dimension can be broadcasted or repeated d_h times along the embedding dimension for element-wise multiplication with the query array with shape (B, N, H, L, d_h). Broadcasting is performed automatically during element-wise multiplication in Step 3.
Just like tied residue-wise attention, we compute the q, k, and v vectors by projecting the MSA feature array with shape (B, N, L, d_msa) with three distinct linear layers. Then, we split the vectors into H d_h-dimensional subvectors by reshaping the array to (B, N, L, H, d_h) and permute so we have a L x d_h matrix of q and v vectors and a transposed d_h x L matrix of k vectors at each head for each sequence in the MSA.
Since there is a one-to-one correspondence between the query vector for position i in sequence n to the (n, i)th sequence weight, we can reduce the computational cost of multiplying every attention weight by its sequence weight by first scaling each d_h-dimensional query vector with a sequence weight. We do this by taking the element-wise product between the sequence weight array with shape (B, N, L, H, 1) broadcasted along the embedding dimension to (B, N, L, H, d_h) and the query array with shape (B, N, H, L, d_h). The result is the L x d_h query matrix where the query vector for position i = [1…L] for sequence n at attention head h is scaled by the (n, i)th sequence weight for head h.
Next, we multiply the scaled query matrix with the transposed key matrix, equivalent to taking the dot product between every pair of query and key vectors to generate an L x L matrix of attention scores for each sequence N.
To get the soft-tied attention scores that will be used to compute attention for all sequences in the MSA, we sum the L x L matrices computed in the previous step from all N sequences.
We can also condense Steps 4 and 5 into a single step with the einsum function.
Since the sequence weights are normalized such that the set of N sequence weights for a single position i add up to 1, we only have to scale each attention score down by √d_h instead of √N*d_h (tied attention).
We apply the softmax function (denoted by SM) across each row of the L x L attention matrix, such that the set of L soft-attention scores for position i attending to every other position in the sequence sum to 1.
The remaining steps are the same as tied multi-head attention. We first use torch.matmul to compute the matrix product of the array of soft-tied attention weights with shape (B, 1, H, L, L) broadcasted to (B, N, H, L, L) and the value array with shape (B, N, H, L, d_h). Then, we permute the dimensions to shape (B, N, L, H, d_h) and concatenate the contextual embeddings across all heads to get shape (B, N, L, d_msa). Finally, we apply a d_msa x d_msa linear transformation layer with learnable weights to process the concatenated embeddings.
The SoftTiedMultiheadAttention module also returns the symmetrized array of soft-tied attention scores which will be used as pair-wise features.
Linearized Attention with Performer
Even though axial attention reduces the receptive field down to only a single row or column, the time and space complexity of computing the attention matrix still grows quadratically as the batch size or the number of sequences in the MSA increases. To reduce the attention mechanism down to linear time, RF uses the Performer architecture by Choromanski et al. which leverages Fast Attention via Positive Orthogonal Random Features (FAVOR+) to linearize the softmax function.
As we described earlier, sequence-wise attention has the following time and space complexities for a single column of the N x L MSA matrix:
Repeating for all L columns of the MSA and B query MSAs in a single training batch, the time and space complexity for the full column-wise mechanism is:
Since the MSAs used in RF have between 2000 to 5000 sequences and a batch size of 64, computing column-wise attention requires significant time and memory.
If we consider the attention mechanism without the nonlinear softmax function, we can reduce the attention mechanism to linear time complexity by leveraging the associative property matrix multiplication.
Instead of explicitly computing the attention matrix A, linearized attention computes the matrix product of the d_msa x N transposed key matrix and the N x d_msa value matrix to get a d_msa x d_msa intermediate matrix. This reduces the time complexity of O(BLNd²) for a single column and O(BLNd²) for the entire MSA array.
Then we compute the matrix product of the N x d_msa query matrix and the d_msa x d_msa intermediate matrix from the previous step to get the final N x d_msa matrix of contextual embeddings which also has reduced time complexity of O(Nd²) for a single column and O(BLNd²) for the entire MSA array.
Since d is much smaller than N (d_msa = 384 in the implementation for RF), the linearized version of sequence-wise attention has much smaller time and space complexity that grows linearly with respect to the number of sequences in the MSA instead of quadratically
However since the softmax function is nonlinear, we cannot write the attention matrix as the matrix product of the query and key matrices.
A method to bypass the need to explicitly compute the attention matrix was introduced by Choromanski et al.with the Performer architecture that leverages Fast Attention Via Positive Orthogonal Random features (FAVOR+) which reduces the attention mechanism to linear time and space complexity.
Linearizing the softmax function with random feature maps, and converting the quadratic time softmax function operating on the N x N attention matrix to a linear time operation. (Source: Alchemy Bio)
At a high level, the Performer architecture converts the original N x d query and key matrices into two transformed N x r matrices Q* and K* constructed with positiveorthogonal random feature projections such that the matrix product is an accurate approximation of the nonlinear softmax kernel applied to the N x N attention matrix A.
For the rest of this section, we will refer to the random feature projections of the query and key vectors/matrix as query* and key* or as Q* and K*.
Then, it leverages the associative property of matrix multiplication to generate the output N x d_h matrix of contextual embeddings without explicitly computing and storing the N x N attention matrix.
Since the normalization step of the softmax function needs the entire row of the attention matrix to ensure that the scores across the row add to 1, we will omit it from our definition of the softmax kernel for now. Later, we will see how this normalization is accounted for.
With this, we define the softmax kernel (SM) as a nonlinear function that takes two vectors q and k, and computes the exponential of the dot product:
We aim to derive a feature mapφ that transforms the d_msa-dimensional query and key vectors into r-dimensional query* and key* vectors such that their dot product approximates the softmax kernel.
Don’t worry if steps feel like magic right now, we will be diving deep into the intuition behind the idea behind the feature map and deriving it from scratch in the next couple of sections. Right now, we just want to gauge a foundational understanding of why we even care to linearize the softmax function in the first place and the end-goal of the feature map φ.
Before focusing specifically on the softmax kernel, let’s take a detour to understand the intuition behind kernel functions in machine learning and the fundamental theorem that allows us to approximate them using random features.
Kernels and Bochner’s Theorem
If you are not familiar with kernel methods, the idea of linearizing a nonlinear function with a feature map may feel unintuitive. Given that kernels are foundational to several domains of machine learning, we will be diving deep into the motivation and theorems that make kernels so powerful, including Bochner’s theorem that directly applies to our construction of the softmax kernel.
Suppose we have a binary classification task and a set of data points in 2-dimensional space that we want to separate with a hyperplane (or straight line in 2D). As you can see, it is impossible to draw a straight line that separates the blue from the red points.
A set of 2-dimensional feature vectors with red and blue labels that are not linearly separable with a linear model (line). (Source)
But as it turns out, we can map each 2-dimensional feature vector into a 3-dimensional vector of new features (referred to as hidden features) using a function φ such that the new set of points in 3D space is linearly separable with a 3D hyperplane. The feature map used in the example below is the degree-2 polynomial feature map, defined as:
The random feature map projects a set of 2-dimensional feature vectors into 3-dimensional hidden feature vectors that are linearly separable. (Source)
Now, we can easily train a simple linear classification model w that classifies the transformed feature vectors φ(x) by computing the dot product, which returns the side (and distance) that the point x lies with respect to the hyperplane w3.
Projecting raw d-dimensional input feature vectors into a higher D-dimensional feature space with a feature map φ and training machine learning models on the set of hidden features is a powerful technique in machine learning.
\(\underbrace{\phi(\mathbf{x}):\mathbb{R}^d\to \mathbb{R}^D}_{\text{project to hidden features}}\;\;\text{then}\;\;\underbrace{\mathbf{w}\cdot \phi(\mathbf{x})}_{\text{train } \mathbf{w}\text{ on hidden features}}\)
However, for tasks that involve higher dimensional features with more complex relationships (unlike the trivial example above), the dimensionality of the hidden feature vectors required to effectively separate the data quickly increases.
This is where kernel functions come in.
Consider the dot product of the 2-dimensional vectors x and z mapped into 3-dimensional space by the degree-2 polynomial kernel above. We can expand the dot product of the 3-dimensional hidden feature vectors as follows:
Essentially, we have shown that the dot product of the 3-dimensional polynomial feature maps can be computed in a lower-dimensional feature space just by squaring thedot product of the original 2-dimensional vectors.
In other words, we can linearly separate our data simply by applying a kernel function that operates in the 2-dimensional feature space without ever having to increase the dimension of our features. Pretty incredible right?
The idea of reducing complex D-dimensional feature maps that capture complex relationships across data into a function that operates directly in the lower d-dimensional space is called the kernel trick.
A kernel, denoted with k, is a linear function that takes two d-dimensional vectors x and z, and returns a scalar value equal to the dot product of a higher D-dimensional hidden feature mapping ofφ(x) and φ(z).
\(\underbrace{k(\mathbf{x,z})}_{\text{kernel}}=\underbrace{\phi(\mathbf{x})\cdot \phi(\mathbf{z})}_{\text{dot product of hidden features}}\)
The degree-2 polynomial kernel generalizes into a set of polynomial kernels of degree p, defined as:
where the dimension of the feature mapping is dependent on the degree of the polynomial p and the dimension of the input feature vectors d4.
\(\underbrace{D=\binom{p+d}{d}}_{\text{dimension of feature map}}\)
In the case of a linear model, the optimal hyperplane w* is a linear combination of the feature vectors x of the training examples, which we can project with the hidden feature map to get φ(x).
By the Representer’s Theorem, the optimal hyperplane is a linear combination of the feature vectors in the training dataset regardless of the feature space that the model is trained in, so we can compute the optimalhyperplane as the hidden feature mappings of a set of n training examples:
\(\mathbf{w}^*=\underbrace{\sum_{i=1}^n\alpha_i\phi(\mathbf{x}_i)}_{\text{weighted sum of input data}}\)
This means that the mechanism of a linear classifier on a feature vector z of a new point can be represented as a linear combination of the kernel function k applied to z with every feature vector x in the training dataset.
\(\begin{align}\underbrace{\mathbf{w}^*\cdot \phi(\mathbf{z})}_{\text{linear classifier}}&=\sum_{i=1}^n\alpha_i\underbrace{\phi(\mathbf{x}_i)\cdot \phi(\mathbf{z})}_{\text{definition of a kernel}}\\&=\sum_{i=1}^n\alpha_ik(\mathbf{x}_i,\mathbf{z})\end{align}\)
How does this relate to approximating the softmax function? Now that we have some intuition on converting feature maps into kernels, we can do the inverse and construct feature maps that compute kernel functions with a dot product.
In fact, Mercer’s theorem states that any positive semi-definite functionk that takes two d-dimensional inputs x and z can be represented as the dot product of a feature mapφof x and z operating in a higher D-dimensional feature space.Note that D is not bounded and can be infinite.
\(\begin{align}\underbrace{k(\mathbf{x,z)}}_{\text{positive-definite function}}=\underbrace{\phi(\mathbf{x})\cdot \phi(\mathbf{z})}_{\text{dot product of feature maps}}\end{align}\)
where the functions k and φ have input and output dimensions:
A function k(x, z) dependent on xand z is positive semi-definite if the integral over all real values of x, zof the function satisfies the following identity:
for all square-integrable functions g. A square-integrable function over all real values is defined as a function where the integral over the square of the function evaluated at all real values x is finite or bounded.
\(\int_{\mathbb{R}}|g(x)|^2dx<\infty\)
All monotonically-increasing functions are not square-integrable since the square of the function approaches infinity as x goes to infinity. In contrast, all probability density functions are square-integrable because the probability over all possible real values must equal 1. Later, we will see that the softmax kernel takes this form.
Let’s break down an example of Mercer’s theorem for a kernel function that has an infinite-dimensional feature mapping: the Radial Basis Function (RBF) kernel.
The RBF kernel is a function that measures the similarity between two d-dimensional vectors x and z by computing the negative squared distance of the features and applying the exponential function.
As the squared distance approaches 0, the exponential approaches 1, and as the squared distance increases, we’re taking the exponential of a large negative value, which approaches 0.
Intuitively, you can also visualize the RBF kernel as a Gaussian probability distribution function centered around x which is close to 1 when z is near x and exponentially falls to 0 as z moves away from x. In RBF kernel methods, this distance function is computed for a new data point z with all training examples x_i to generate a prediction for the label of z based on the distances and labels of the training examples.
An RBF kernel is like a Gaussian probability distribution function centered around each training data point x. The value of the RBF kernel for a point z is determined by its distance from x. (Source)
σ determines how quickly the value falls to 0, or the variance of the Gaussian. This determines the length scale of the training data that a new data point considers when making a prediction. We will assume σ = 1 from now on.
We can decompose the numerator of the RBF kernel to write it in terms of a dot product:
Substituting this into the original equation, we have:
\(\begin{align}k_{\text{RBF}}(\mathbf{x}, \mathbf{z})&=\exp\left(-\frac{||\mathbf{x}||^2-2\left(\mathbf{x \cdot z}\right)+||\mathbf{z}||^2}{2}\right)\\&=\exp\left(\frac{-||\mathbf{x}||^2}{2}+\left(\mathbf{x \cdot z}\right)-\frac{||\mathbf{z}||^2}{2}\right)\\&=\underbrace{\exp\left(\frac{-||\mathbf{x}||^2}{2}\right)}_{\text{only dependent on }\mathbf{x}}\exp(\mathbf{x}\cdot \mathbf{z})\underbrace{\exp\left(\frac{-||\mathbf{z}||^2}{2}\right)}_{\text{only dependnet on }\mathbf{z}}\end{align}\)
Since the first and last terms are only dependent on one of the feature vectors, we can consider them as constants used to scale the inputs of the dot product. The term that we have to worry about is exponential of the dot product between x and z.
Recall that the Taylor expansion of the exponential function is given by:
\(\exp(x)=e^{x}=\sum_{n=1}^\infty\frac{x^n}{n!}\)
Using the Taylor expansion of the exponential, we can write the exponential of the dot product between x and z as:
By definition, the sum of two kernels is another kernel:
\(\begin{align}\underbrace{k_1(\mathbf{x,z})+k_2(\mathbf{x,z})}_{\text{sum of two kernels}}&=\phi_1(\mathbf{x})\cdot \phi_1(\mathbf{z})+\phi_2(\mathbf{x})\cdot \phi_2(\mathbf{z})\\&=\begin{bmatrix}\phi_1(\mathbf{x})\\\phi_1(\mathbf{x})\end{bmatrix}\cdot \begin{bmatrix}\phi_2(\mathbf{z})\\\phi_2(\mathbf{z})\end{bmatrix}\\&=\phi_3(\mathbf{x})\cdot \phi_3(\mathbf{z})\\&=\underbrace{k_3(\mathbf{x,z})}_{\text{another kernel}}\end{align}\)
It follows that the dimension of the feature map for the combined kernel is the sum of the two original kernels:
Therefore, the RBF kernel is indeed a kernel which is the infinite sum of the polynomial kernels of degrees r, where r goes to infinity.
\(\begin{align}k_{\text{RBF}}(\mathbf{x,z})&=\underbrace{\exp\left(\frac{-||\mathbf{x}||^2}{2}\right)\exp\left(\frac{-||\mathbf{z}||^2}{2}\right)}_{\text{constant }C}\sum_{r=1}^\infty\frac{k^r_{\text{poly}}(\mathbf{x,z})}{r!}\\&=C\underbrace{\sum_{r=1}^\infty\frac{k^r_{\text{poly}}(\mathbf{x,z})}{r!}}_{\text{infinite sum of poly kernels }}\end{align}\)
As the degree of the polynomial increases to infinity, the dimension of the D-dimensional random feature map also approaches infinity. Therefore, the dimension of the feature map for the RBF kernel is infinite.
Since it is impossible to compute an infinite-dimensional feature map, we can only approximate the RBF kernel with a finite-dimensional feature map. To construct a feature map that produces an accurate approximation, we need to discuss another fundamental theorem: Bochner’s theorem.
Bochner’s theorem states that all positive semi-definite, shift-invariant kernels can be written as the Fourier transform of a probability distributionp(ω).
A shift invariant kernel is defined as a kernel that depends only on the displacement between two vectors and not their exact coordinates.
\(\underbrace{k(\mathbf{x-z})}_{\text{depends only on displacement}}=k\underbrace{\bigg(\mathbf{(x+y)-(z+y)}\bigg)}_{\text{displacement is equal}}\)
To understand Bochner’s theorem, we need to first understand the Fourier transform. Although several incredible resources5 exist that break down this concept, I found it worthwhile to put it into my own words to lead into our discussion on Bochner’s theorem.
First, we will break down the Fourier series decomposition of periodic functions and transition into the Fourier transform of non-periodic functions
By Euler’s formula6, we can write any sinusoidal function as the complex exponential function that defines a point on the unit circle, where i is the complex number, x is the time variable, 2π is a single rotation over the unit circle in radians, and k/L is the frequency.
The Fourier series allows us to decompose any periodic function as the discrete sum of sinusoidal functions of specific frequencies. Suppose we have a periodic function over an interval of length L from -L/2 to L/2. Then, we can write it as an discrete sum of different frequencies k/L or equivalently as the discrete sum of different periods (inverse of frequency) L/k such that there is an integer number of oscillations k over the interval L.
We can rewrite the equation above in terms of the angular frequency ω that defines number of rotations over the unit circle with units of radians per second.
Therefore, any sinusoidal functions can be expressed as a discrete sum of frequencies of angular frequencies ranging from negative to positive infinity.
But what exactly are the Fourier coefficients? Intuitively, the Fourier coefficients determine how much of the function f(x) is defined by the sinusoidal function of each angular frequency. To derive the expression for c_k, we will show that the exponential functions at any two different angular frequencies k and l are orthogonal, and thus the set of expoenntial functions for all k from negative to positive infinity form a orthogonal basis of the frequency domain.
By definition, the inner product of two complex-valued functions is the integral of the product of the first function and the complex conjugate of the second function over a specified interval7. Since we have defined the interval of our periodic function to be -L/2 to L/2, we can write:
\(\scriptsize\begin{align}&=\frac{1}{i(\omega_k-\omega_l)}\bigg[\exp\left(\frac{iL}{2}\left(\frac{2\pi (k-l)}{L}\right)\right)-\exp \left(-\frac{iL}{2}\left(\frac{2\pi (k-l)}{L}\right)\right)\bigg]\\&=\frac{1}{i(\omega_k-\omega_l)}\bigg[\exp\big(i\pi (k-l)\big)-\exp\big(-i\pi (k-l)\big)\bigg]\\&=\frac{1}{i(\omega_k-\omega_l)}\underbrace{\bigg[\underbrace{\cos\big(\pi (k-l)\big)}_{=-1 \text{ or } 1}+i\underbrace{\sin\big(\pi (k-l)\big)}_{=0}-\underbrace{\cos\big(-\pi (k-l)\big)}_{\text{=-1 or 1}}-i\underbrace{\sin\big(-\pi (k-l)\big)}_{\text{=0}}\bigg]}_{\text{cancels to }0 \text{ since sine and cosine evaluate to either 1 or -1 and 0 for any integer multiple of }\pi}\\&=0\end{align}\)
Therefore, we’ve demonstrated that the exponential functions at any two different angular frequencies k and l are orthogonal. However, something interesting happens when k = l.
\(\begin{align}\big\langle \exp(i\omega_kx), \exp(i\omega_lx)\big\rangle &=\int_{-L/2}^{L/2}\exp(ix\underbrace{(\omega_k-\omega_l)}_{\text{0 when }k=l})dx\\&=\int_{-L/2}^{L/2}\exp(0)dx\\&=\int_{-L/2}^{L/2}1dx\\&=\frac{L}{2}+\frac{L}{2}\\&=L \end{align}\)
We have shown above that the set of complex exponentials for all integer number of oscillations k and l on the interval of length L are mutually orthogonal to every other function in the set except for itself.
\(\big\langle \exp(i\omega_kx), \exp(i\omega_lx)\big\rangle=\begin{cases}0&k\neq l \\L&k=l\end{cases}\)
Given that the set of exponential functions of each angular frequencyω_k form an orthogonal basis of all periodic functions over -L/2 to L/2, we can determine how much of the function f(x) is defined by each frequency simply by projecting the function f(x) onto the orthogonal basis function like we would do in vector space, which become our Fourier coefficients.
\(\begin{align}\underbrace{\big\langle f(x), \exp(i\omega_kx)\big\rangle }_{\text{projection of }f(x)\text{ onto basis function}}&=\int_{-L/2}^{L/2}f(x)\underbrace{\exp(-i\omega_kx)}_{\text{complex conjugate}}dx\\\end{align}\)
When projecting a vector v onto a basis vector u, we must divide the basis vector by its norm such that the dot product is the component of v in the direction of u. Similarly, when projecting functions onto a basis function, we divide by the norm of the basis function which is equal to the inner product of the function with itself. Since the norm of the complex exponential functions is equal to L, our Fourier coefficients are given by:
But how do we extend this idea from periodic functions to non-periodic functions like kernel functions? It turns out we can simply compute the Fourier series decomposition for the periodic function bounded by -L/2 to L/2 for the limit as L goes to infinity, or in other words, as the period of the sinusoid grows to infinity.
In the Fourier series, we compute the infinite sum of discrete values of ω_k where k is an integer that increments by 1. Therefore, the change in ω with each successive value of k is given by:
\(\Delta \omega = \frac{2\pi \Delta k}{L}=\frac{2\pi}{L}\tag{$\Delta k =1$}\)
Since the change in angular frequency is inversely proportional to L, taking the limitas L goes to infinity is equivalent to taking the limit as the change in angular frequency goes to 0. In other words, the Fourier transform treats angular frequency as a continuous variable instead of a set of discrete variables for integer values of k.
\(L\to \infty\implies \Delta \omega \to 0\)
Therefore, we can write the limit in terms of angular frequency to get:
\(\begin{align}f(x)&=\lim_{\Delta \omega\to 0}\sum_{k=-\infty}^{\infty}\underbrace{\left(\frac{\Delta \omega}{2\pi }\int_{\frac{-\pi}{\Delta \omega}}^{\frac{\pi}{\Delta \omega}}f(x)\exp(-i\omega x)dx\right)}_{c_k}\exp\left(i\omega x\right)\\&=\lim_{\Delta \omega\to 0}\sum_{k=-\infty}^{\infty}\underbrace{\left[\left(\frac{1}{2\pi }\int_{\frac{-\pi}{\Delta \omega}}^{\frac{\pi}{\Delta \omega}}f(x)\exp(-i\omega x)dx\right)\exp\left(i\omega x\right)\right]\Delta\omega}_{\text{moving }\Delta \omega\text{ outside the bracket to get a Riemann sum}} \end{align}\)
We can observe that the limit shown above is in the form of a Riemann sum from -∞ to +∞, which we can simply write as the integral from -∞ to +∞ with respect to infinitesimal changes in ω.
\(\begin{align}f(x)&=\int_{-\infty}^{\infty}\left(\frac{1}{2\pi }\underbrace{\int_{-\infty}^{\infty}f(x)\exp(-i\omega x)dx}_{\text{limits are infinity as }\Delta\omega \to 0}\right)\exp\left(i\omega x\right)\underbrace{d\omega}_{\text{infinitesimal change}} \end{align}\)
It turns out that the inner integral is exactly our definition of the inner product between the function f(x) and the exponential function.
\(\begin{align}f(x)&=\int_{-\infty}^{\infty}\left(\frac{1}{2\pi }\underbrace{\int_{-\infty}^{\infty}f(x)\exp(-i\omega x)dx}_{\text{definition of inner product!}}\right)\exp\left(i\omega x\right)d\omega \\&=\int_{-\infty}^{\infty}\underbrace{\frac{1}{2\pi }}_{\text{constant}}\underbrace{\big\langle f(x), \exp(i\omega x)\big\rangle}_{\text{Fourier transform of }f(x)} \exp\left(i\omega x\right)d\omega \end{align}\)
But now since ω is a continuous variable from -∞ to +∞, the inner product is a continuous distribution of projections of f(x) onto the infinite frequency space of sinusoidal functions with angular frequenciesω = -∞ to +∞. We will define this inner product as the Fourier transform p(ω) of f(x) or the continuous distribution of frequencies that compose f(x). Just like the Fourier coefficients, the Fourier transform is scaled down by the norm of the complex exponential.
Since p(ω) is also just an infinite integral of a function f(x) projected over the orthogonal basis of exponential functions (the negative sign just indicates rotation clockwise over the unit circle rather than counterclockwise), it is just another continuous non-periodic function like f(x) except over the frequency domain ω instead of the “time” domain x.
The inverse Fourier transform is the function f(x) represented as an integral (or continuous sum) of a distribution function p(ω) over the space of angular frequencies ω. In other words, f(x) is the inverse Fourier transform of p(ω).
\(\begin{align}f(x)&=\int_{-\infty}^{\infty}\underbrace{p(\omega)}_{\text{distribution of }\omega} \exp\left(i\omega x\right)d\omega \end{align}\)
Intuitively, p(ω) is like a probability distribution indicating the probability of each angular frequency ω occurring in the decomposition of f(x); but as with all probability distributions, the actual probability of f(x) at a particular value of ω is 0 since the integral at a single point is undefined.
Now that we understand the background of the Fourier transform, we can deconstruct the meaning of Bochner’s theorem.
Bochner’s theorem states that all positive semi-definite, shift-invariant kernels can be represented as an inverse Fourier transform over the distribution of angular frequencies defined byp(ω). Since kernel functions operate on d-dimensional vectors, we can extend the probability distribution of angular frequencies to be a function over a d-dimensional vector space of angular frequencies.
Instead of operating in the time domain, the kernel operates over the vector domain. Therefore, the integral from -∞ to +∞ is equivalent to taking the integral of all real-valued realizations of the d-dimensional vector of angular frequencies ω.
By definition, the expected value E[X(ω)] of a function X over the continuous random variable ω is given by the probability of a specific value of ω being sampled from p(ω) multiplied by X(ω) over all possible real values for ω.
Intuitively, the expected value is the value that the function X(ω) converges to after infinite samples of ω.
Therefore, we see that Bochner’s theorem can be written as the expected value of the exponential function over all possible angular frequencies ω drawn from p(ω).
\(\begin{align}k(\mathbf{x-y})&=\underbrace{\int _{\omega\in \mathbb{R}^d}\underbrace{p(\vec{\omega})}_{\text{probability distribution of }\omega}\underbrace{\exp\bigg(i\vec{\omega}\cdot (\mathbf{x-z})\bigg)}_{\text{function dependent on random var }\omega}d\vec{\omega}}_{\text{expected value of the function }\exp\big(i\vec{\omega}\cdot (\mathbf{x-y})\big)}\\&=\underbrace{\mathbb{E}_{\vec{\omega}\sim p(\vec{\omega})}\bigg[\exp\big(i\vec{\omega}\cdot (\mathbf{x-z)}\big)\bigg]}_{\text{expected value over all }\omega\text{ sampled from }p(\omega)}\end{align}\)
Since it is impossible to sample all possible realizations of ω from p(ω), we can approximate the expected value by sampling a discrete number of angular frequencies and taking the average over the number of samples.
Intuitively, we are projecting the input vector (x - z) onto an angular frequency vector ω drawn from the Fourier transform p(ω) and then taking the average of all the periodic functions with each frequency to estimate the infinite sum of periodic functions for angular frequencies over the distribution p(ω).
Since we are only working with real-valued kernel functions over a real-valued probability distributionp(ω), we can replace the complex exponential with only the real-valued cosine component of Euler’s formula.
Using trigonometric identities, we can separate the exponential function as the product of two exponentials dependent only on either x or z and then convert it into a single dot product between two random feature maps of the original vectors x and z.
Now we observe that the sum of the dot product over different values ofω_k can simply be written as a single dot product of the cosine/sine vectors for different values of ω_k concatenated into a single 2r-dimensional vector.
Therefore, we have derived a finite-dimensional random feature map that approximates the feature map of any shift-invariant kernels given some distribution p(ω). This allows us to approximate kernels with infinite-dimensional feature maps like the RBF kernel with a finite number of random feature projections ω.
Intuitively, each random feature vector ω as a d-dimensional vector pointing in a random direction to a point sampled from the Fourier transform p(ω). Taking its dot product with xcomputes the magnitude of x in the direction of ω. Then, by applying the cosine and sine functions to each magnitude, we map to a point on the unit circle. After projecting x onto multiple random directions ω and mapping to the unit circle, we obtain multiple points on the unit circle representing the vector x.
The random feature map projects x onto r different directions for ω and maps the magnitude of each projection onto a point on the unit sphere. (Source: Alchemy Bio)
Repeating for the vector z, we obtain another unit circle with points representing z. Taking the dot product between the mapping of x and the mapping of z essentially computes the similarity of each pair of coordinates corresponding to a single direction of ω and summing across all directions ω sampled from the Fourier transform p(ω). The resulting sum is an unbiased estimator of the the kernel k.
Alternatively, you can imagine of each vector ω as evaluating the relationship between x and z in a way that mimics a single component of the kernel function, and repeating for multiple ω, we reach a more accurate representation of the kernel.
But what exactly is the function p(ω) and how does it relate to the kernel?
It turns out that just by changing the probability distribution p(ω), we can approximate completely different kernels using the same trigonometric Fourier random feature maps defined above.
Kernels and the probability distributions p(ω) that can be used to generate the random feature map. (Source)
In the next section, we will walk through the derivation for the distribution p(ω) that approximates the softmax kernel, and realize that it is just the Gaussian distribution.
Trigonometric Random Feature Estimator
We defined the softmax kernel (SM) as the exponential of the dot product between the query and key vectors.
Since Bochner’s theorem applies only to shift-invariant kernels and the dot product depends on the magnitude and direction of each vector, we need to decompose the softmax function to depend on the difference between q and k.
Recall from earlier that the squared norm of the difference between two vectors can be decomposed into three terms.
Substituting this definition of the RBF kernel into the softmax kernel, we get an estimator for the softmax kernel (denoted with the hat on ‘SM’ and subscript r correspondent the number of random features).
However, we still have yet to define the probability function p(ω) that the random feature vectors are sampled from. To derive this function, let’s recall the definition of a Fourier transform for a scalar function f.
Translating this definition to the kernel function k operating on d-dimensional vectors, we take the integral over the d-dimensional vector space and divide by the norm of the scalar exponential raised to the power of d since the inner product of the two functions is evaluated for all d entries of the input vector. For clarity, we let Δ = q - k for the rest of the derivation.
We aim to reduce this function p(ω) such that it is dependent only on ω so that we can sample our random feature vectors from this distribution independent from the inputs to the kernel. To do this, let’s expand the dot product inside the second exponential using the same technique as before:
Substituting this expression into the exponential function, we have:
\(\small\begin{align}p(\vec{\omega})&=\frac{1}{(2\pi)^d}\int _{\mathbb{R}^d}\exp\left(\frac{-||\vec{\Delta}||^2}{2}\right)\exp\left(\frac{||\vec{\omega}||^2}{2}+\frac{||\vec{\Delta}||^2}{2}-\frac{||-i\vec{\omega}-\vec{\Delta}||^2}{2}\right)d\vec{\Delta}\\&=\frac{1}{(2\pi)^d}\int _{\mathbb{R}^d}\underbrace{\exp\left(\frac{-||\vec{\Delta}||^2}{2}\right)\exp\left(\frac{||\vec{\Delta}||^2}{2}\right)}_{\text{cancels out to 1}}\exp\left(\frac{||\vec{\omega}||^2}{2}-\frac{||-i\vec{\omega}-\vec{\Delta}||^2}{2}\right)d\vec{\Delta}\end{align}\)
We can cancel out some terms to get a simplified form:
Now, we can observe that the expression inside the integral resembles a Gaussian distribution. To eliminate the dependence on Δ, let’s compute the integral of the d-dimensional multivariate Gaussian and use our result to reduce the equation above.
With a bit of calculus8, we can find that the integral of the 1-dimensional Gaussian distribution is given by:
For a d-dimensional multivariate Gaussian, we can expand the squared norm of the input d-dimensional vector δ as the sum of all d squared entries. Then, we separate the integral for each variable δ_1, …, δ_d to generalize the integral of the 1-dimensional Gaussian distribution to the d-dimensional case.
Therefore, the normalized d-dimensional Gaussian distribution with an integral of 1 (area under the distribution or total probability is 1) is obtained by dividing by the integral obtained above.
\(\int_{\mathbb{R}^d}\underbrace{(2\pi)^{\frac{-d}{2}}\exp\left(-\frac{||\vec{\delta}||^2}{2}\right)}_{\text{normalized Gaussian probability function }\mathcal{N}(\vec{0}_d,\mathbf{I}_d)}d\vec{\delta}=1\)
Since the term inside the integrand is just a d-dimensional Gaussian distribution with a mean of -iω with unit variance and the integral does not change when shifting the mean, we can reduce the integral over Δ as the integral of a d-dimensional Gaussian.
\(\begin{align}p(\vec{\omega})&=\frac{1}{(2\pi)^d}\exp\left(\frac{||\vec{\omega}||^2}{2}\right)\underbrace{\int _{ \mathbb{R}^d}\exp\left(\frac{-||-i\vec{\omega}-\vec{\Delta}||^2}{2}\right)d\Delta}_{(2\pi)^{\frac{d}{2}}}\\&=\frac{1}{(2\pi)^d}\exp\left(\frac{||\vec{\omega}||^2}{2}\right)(2\pi)^{\frac{d}{2}}\\&=(2\pi)^{\frac{d}{2}-d}\exp\left(\frac{||\vec{\omega}||^2}{2}\right)\\&=\underbrace{(2\pi)^{\frac{-d}{2}}\exp\left(\frac{||\vec{\omega}||^2}{2}\right)}_{\text{just a Gaussian centered at 0}}\end{align}\)
By our earlier definition of the normalized Gaussian, we have shown that the Fourier transformp(ω) of the RBF kernel is just a d-dimensional multivariate Gaussian centered at the origin with unit variance.
Since the co-variance of the Gaussian distribution is 0 (non-diagonal entries of the covariance matrix are zero), the entries of ω are independent we can approximate the RBF kernel by sampling each entry of each d-dimensional random feature vector ωidentically andindependently from the 1-dimensional Gaussian distribution with a mean of 0 and a variance of 1.
\(\vec{\omega} \overset{\mathrm{iid}}{\sim}\mathcal{N}\left(\begin{bmatrix}0\\0\\\vdots\\0\end{bmatrix},\underbrace{\begin{bmatrix}1&0&\dots&0\\0&1&\dots &0\\\vdots&\vdots&\ddots&\vdots\\0&0&\dots &1\end{bmatrix}}_{\text{covariance matrix is identity }\mathbf{I}_d}\right)\)
Therefore, the expanded inverse Fourier transform of the RBF kernel is given by:
In expectation, the RBF kernel converges to the complex exponential of q - k for infinite samples of ω over the Gaussian distribution.
\(\begin{align}k_{\text{RBF}}(\mathbf{q-k})&=\underbrace{\exp\left(\frac{-||\mathbf{q-k}||^2}{2}\right)}_{\text{definition of RBF kernel}}\\&=\underbrace{\mathbb{E}_{\vec{\omega}\sim \mathcal{N}(\vec{0}, \mathbf{I}_d)}\bigg[\exp\big(i\vec{\omega}\cdot (\mathbf{q-k)}\big)\bigg]}_{\text{expected value of infinite random feature map}}\\&\approx\underbrace{ \frac{1}{r}\sum_{k=1}^r\exp\big(i\vec{\omega_k}\cdot (\mathbf{q-k)}\big)}_{\text{estimator with finite-dim random feature map}}\tag{$\vec{\omega}\sim \mathcal{N}(\vec{0}, \mathbf{I}_d)$}\end{align}\)
Substituting the RBF kernel into the equation for the true softmax kernel, we get:
By drawing r discrete d-dimensional random feature vectors from the Gaussian distribution and taking the average of the complex exponential evaluated with each random feature, we get an unbiased estimator for the softmax kernel (denote with the hat on ‘SM’ and subscript r which is the number of features)
Earlier, we defined the trigonometric estimator of the softmax function by replacing the complex exponential with only the real cosine component and expanding using trigonometric identities. Now that we know p(ω), we can draw our samples from the Gaussian distribution to compute an approximation of the softmax kernel with the trigonometric estimator.
\(\begin{align}\underbrace{\widehat{\text{SM}}_r^{\text{trig}}(\mathbf{q,k})}_{\text{trig estimator}}&=\exp\left(\frac{||\mathbf{q}||^2}{2}\right)\exp\left(\frac{||\mathbf{k}||^2}{2}\right) \frac{1}{r}\sum_{k=1}^r\cos\big(\vec{\omega_k}\cdot (\mathbf{x-z)}\big)\\&=\exp\left(\frac{||\mathbf{q}||^2}{2}\right)\exp\left(\frac{||\mathbf{k}||^2}{2}\right) \underbrace{\frac{1}{r}\sum_{k=1}^r\left(\begin{bmatrix}\cos(\vec{\omega_k}\cdot \mathbf{x})\\\sin(\vec{\omega_k}\cdot \mathbf{x})\end{bmatrix}\cdot \begin{bmatrix}\cos(\vec{\omega_k}\cdot \mathbf{z})\\\sin(\vec{\omega_k}\cdot \mathbf{z})\end{bmatrix}\right)}_{\text{average of }r\text{ samples from }\vec{\omega}\sim\mathcal{N}(\vec{0}, \mathbf{I}_d)}\end{align}\)
which can be expanded into the dot product of two random feature maps:
To evaluate whether this estimator is an accurate approximation of the softmax function given random samples of ω, let’s derive the mean-squared error.
By definition, the mean-squared error (MSE) of an estimator that maps data (query and key embeddings) to a quantity (attention score) is given by the following equation:
\(\text{MSE}(\hat{\theta})=\underbrace{\mathbb{E}_{\theta}\left[(\hat{\theta}-\theta)^2\right]}_{\text{expected squared error of estimator }\hat{\theta}\text{ over the unknown parameter }\theta}\)
For an unbiased estimator, the estimated value is equal to the unknown parameter, meaning that as the number of samples increases to infinity, the average of all the estimates will always converge to the unknown parameter.
\(\begin{align}\text{MSE}(\hat{\theta})&=\underbrace{\mathbb{E}_{\theta}\left[\left(\hat{\theta}-\mathbb{E}_{\theta}[\hat{\theta}]\right)^2\right]}_{\text{definition of variance}}\\&=\mathbb{E}_{\theta}\big[\hat{\theta}^2\big]-\left(\mathbb{E}_{\theta}[\hat{\theta}]\right)^2\\&=\text{Var}(\hat{\theta})\end{align}\)
Therefore, the MSE of an unbiased estimator is equal to the variance of the estimator. By definition, the variance of a random variable is equal to the expected value of the squared distance between the random variable and the mean.
Since the trigonometric random feature estimator is unbiased, we can compute the MSE by computing its variance.
When a random variable X is scaled by a constantc, the variance of the scaled random variable cX is equal to the constant squared multiplied by the variance of the random variable X.9
\(\text{Var}[cX]=c^2\text{Var}[X]\)
Therefore, we can take the constant terms that are not dependent on ω out of the variance and square them to get:
\(\begin{align}\small\text{MSE}\bigg(\widehat{\text{SM}}^{\text{trig}}_r(\mathbf{q},\mathbf{k})\bigg)&=\text{Var}\left[\underbrace{\exp\left(\frac{||\mathbf{q}||^2+||\mathbf{k}||^2}{2}\right)\frac{1}{r}}_{\text{constant (not dependent on }\omega)}\underbrace{\sum_{k=1}^r\exp\big(\vec{\omega}_k\cdot (\mathbf{q}-\mathbf{k})\big)}_{\text{random variable}}\right]\nonumber\\&=\frac{1}{r^2}\underbrace{\exp\left(\frac{||\mathbf{q}||^2+||\mathbf{k}||^2}{2}\right)^2}_{\exp(x)^2=(e^x)^2=e^{2x}}\text{Var}\left[\sum_{k=1}^r\exp\big(\vec{\omega}_k\cdot (\mathbf{q}-\mathbf{k})\big)\right]\\&=\frac{1}{r^2}\underbrace{\exp\left(\frac{2(||\mathbf{q}||^2+||\mathbf{k}||^2)}{2}\right)}_{\text{cancel out 2}}\text{Var}\left[\sum_{k=1}^r\exp\big(\vec{\omega}_k\cdot (\mathbf{q}-\mathbf{k})\big)\right]\\&=\frac{1}{r^2}\exp\left(||\mathbf{q}||^2+||\mathbf{k}||^2\right)\text{Var}\left[\sum_{k=1}^r\exp\big(\vec{\omega}_k\cdot (\mathbf{q}-\mathbf{k})\big)\right]\end{align}\)
Bienaymé’s identity states that the variance of the sum of uncorrelated random variables is equal to the sum of the variances of those same variables. A pair of random variables are uncorrelated when their covariance matrix is 0 and there is no linear dependence between them. Since independence implies uncorrelation, this identity also holds for independent random variables, where the probabilities of each random variable do not influence the other.
We can strengthen this identity to state that the sum of independently and identically random variables, which share the same variance is equal to the variance multiplied by the number of summations:
\(\begin{align}\text{Var}\left[\sum_{k=1}^rX_k\right]&=\underbrace{\sum_{k=1}^r\text{Var}\left[X_k\right]}_{\text{if variance is equal for all }k}\\&=r\text{Var}[X]\end{align}\)
Since the random feature vectors are independently and identically distributed over the multivariate Gaussian, we can use Bienaymé’s identity to simplify the variance of the sum over all r random variables.
\(\small\begin{align}\text{MSE}\bigg(\widehat{\text{SM}}^{\text{trig}}_r(\mathbf{q},\mathbf{k})\bigg)&=\frac{1}{r^2}\exp\left(||\mathbf{q}||^2+||\mathbf{k}||^2\right)\underbrace{\text{Var}\left[\sum_{k=1}^r\exp\big(\vec{\omega}_k\cdot (\mathbf{q}-\mathbf{k})\big)\right]}_{\text{variance of sum = sum of variances}}\\&=\frac{1}{r^2}\exp\left(||\mathbf{q}||^2+||\mathbf{k}||^2\right)\underbrace{\sum_{k=1}^r\text{Var}\bigg[\exp\big(\vec{\omega}_k\cdot (\mathbf{q}-\mathbf{k})\big)\bigg]}_{\omega\text{ have equal variance for all }k}\\&=\underbrace{\frac{1}{r^2}\exp\left(||\mathbf{q}||^2+||\mathbf{k}||^2\right)r}_{\text{cancel out }r}\text{Var}\bigg[\exp\big(\vec{\omega}\cdot (\mathbf{q}-\mathbf{k})\big)\bigg]\\&=\frac{1}{r}\exp\left(||\mathbf{q}||^2+||\mathbf{k}||^2\right)\text{Var}\bigg[\exp\big(\vec{\omega}\cdot (\mathbf{q}-\mathbf{k})\big)\bigg]\end{align}\)
We will now expand the variance term before substituting it back into our equation. Using our definition of variance as the expectation of the squared random variable subtracted by the squared of the expectation, we have:
Let’s break down the first estimated value term labeled (1).
For this step, we will need the squared cosine trigonometric identity:
\(\cos^2(x)=\frac{1-\cos(2x)}{2}\)
We will also be using the some properties of expected value. First, linearity of expectation is a property that states the expected value of the sum of random variables is equal to the sum of the expected values of the variables. This holds for the sum of any set of variables, even if they are not independent or uncorrelated. Second, the expectation of a constant is the constant itself. And third, it the expected value of a random variable is scaled by a constant is equal to the constant multiplied by the expected value.
From our earlier derivation, we found that the estimated value of the infinite-dimensional random feature map of the trigonometric estimator is equal to the RBF kernel.
\(\begin{align}\mathbb{E}\bigg[\cos^2\big(\vec{\omega}\cdot (\mathbf{q-k})\big)\bigg]&=\frac{1}{2}-\frac{1}{2}\mathbb{E}\left[\cos\big(\vec{\omega}\cdot \underbrace{2(\mathbf{q-k})}_{\text{scale delta by 2}}\big)\right]\\&=\frac{1}{2}-\frac{1}{2}\underbrace{\exp\left(\frac{-||2(\mathbf{q}-\mathbf{k})||^2}{2}\right)}_{\text{RBF kernel for }2\mathbf{q}\text{ and }2\mathbf{k}}\\&=\frac{1}{2}-\frac{1}{2}\exp\underbrace{\left(\frac{-4||\mathbf{q}-\mathbf{k}||^2}{2}\right)}_{\text{square constant}}\\&=\frac{1}{2}-\frac{1}{2}\exp\left(-2||\mathbf{q}-\mathbf{k}||^2\right)\end{align}\)
We can also rewrite the second expected value term labeled (2) as:
Since the value of the RBF kernel is bounded between (0,1], where 1 indicates that the vectors are the same, and as the vectors become farther apart, the RBF kernel exponentially approaches 0. Analyzing the equation for MSE, we observe that for the limit as the RBF kernel approaches 0, the MSE grows exponentially as a function of the squared norm of the query and key vectors.
\(\small\begin{align}\lim_{k_{\text{RBF}}\to 0}\bigg(\small\text{MSE}\bigg(\widehat{\text{SM}}^{\text{trig}}_r(\mathbf{q},\mathbf{k})\bigg)\bigg)&=\lim_{k_{\text{RBF}}\to 0}\bigg(\frac{1}{2r}\exp\left(||\mathbf{q}||^2+||\mathbf{k}||^2\right)\bigg(1-k_{\text{RBF}}(\mathbf{q-k})\bigg)^2\bigg)\\&=\frac{1}{2r}\exp\left(||\mathbf{q}||^2+||\mathbf{k}||^2\right)\bigg(1-0\bigg)^2\\&=\underbrace{\frac{1}{2r}\exp\left(||\mathbf{q}||^2+||\mathbf{k}||^2\right)}_{\text{when vectors are far apart, MSE increases exponentially }}\end{align}\)
Since we know that the softmax function decreases as the RBF kernel decreases, we can guess that MSE also increases for small values of the true softmax kernel. To confirm our intuition, we can write the RBF kernel in terms of the true softmax function:
As the true value of the softmax function approaches 0, the MSE of the positive random feature estimator increases exponentially to infinity. Therefore, the trigonometric random feature estimator is highly inaccurate in estimating small attention scores.
To overcome the inaccuracy of trigonometric random features, the Performer architecture leverages positive random feature maps.
Positive Random Feature Estimator
The Performer architecture uses positive random features instead of cosine/sine features used to approximate the softmax kernel. Since attention scores are all positive values between 0 and 1, negative values generated from sine/cosine random features result in inaccurate approximations, especially for small attention scores.
Since all attention scores are positive values between 0 and 1, Choromanski et al. found that the negative values generated from cosine/sine functions led to inaccurate approximations, especially for low attention scores that are common in long-range attention mechanisms.
Therefore, they proposed a positive random feature map for the softmax function. To understand the origin of the positive random feature map, let’s consider the complex-valued feature map for the softmax kernel.
Notice that if we removed the complex number i, the exponential function would always evaluate to a positive value. Since the product of a function and its complex conjugate is analogous to the product for non-complex functions, we can convert the feature map into the real-valued exponential function and see if we can derive the softmax kernel for some probability distribution p(ω).
\(\small\begin{align}\mathbb{E}_{\vec{\omega}\sim p(\vec{\omega})}\bigg[\exp(\vec{\omega}\cdot \mathbf{x})\exp(\vec{\omega}\cdot\mathbf{z})\bigg]&=\int_{\mathbb{R}^d}p(\vec{\omega})\exp\bigg(\frac{||\vec{\omega}||^2}{2}+\frac{||\mathbf{q+k}||^2}{2}-\frac{||\vec{\omega}-(\mathbf{q}+\mathbf{k})||^2}{2}\bigg)d\vec{\omega}\\&=\int_{\mathbb{R}^d}p(\vec{\omega})\exp\bigg(\frac{||\vec{\omega}||^2}{2}\bigg)\underbrace{\exp\bigg(\frac{||\mathbf{q+k}||^2}{2}\bigg)}_{\text{not dependent on }\vec{\omega}}\exp\bigg(-\frac{||\vec{\omega}-(\mathbf{q}+\mathbf{k})||^2}{2}\bigg)d\vec{\omega}\end{align}\)
Since the second term is not dependent on ω, we can take it out of the integral:
\(\small\begin{align}\mathbb{E}_{\vec{\omega}\sim p(\vec{\omega})}\bigg[\exp(\vec{\omega}\cdot \mathbf{x})\exp(\vec{\omega}\cdot\mathbf{z})\bigg]&=\exp\bigg(\frac{||\mathbf{q+k}||^2}{2}\bigg)\underbrace{\int_{\mathbb{R}^d}p(\vec{\omega})\exp\bigg(\frac{||\vec{\omega}||^2}{2}\bigg)\exp\bigg(-\frac{||\vec{\omega}-(\mathbf{q}+\mathbf{k})||^2}{2}\bigg)d\vec{\omega}}_{\text{needs to cancel to 1}}\end{align}\)
Since the expected value should be independent of our random variable ω, we need to define p(ω) such that the integral cancels to 1.
Recall that the integral of the normalized multi-variate Gaussian evaluates to 1:
\(\int_{\mathbb{R}^d}\underbrace{(2\pi)^{\frac{-d}{2}}\exp\left(-\frac{||\vec{\delta}||^2}{2}\right)}_{\text{normalized Gaussian probability function }\mathcal{N}(\vec{0}_d,\mathbf{I}_d)}d\vec{\delta}=1\)
To get this result, we need to define p(ω) such that:
\(\begin{align}p(\vec{\omega})\exp\bigg(\frac{||\vec{\omega}||^2}{2}\bigg)&=(2\pi)^{\frac{-d}{2}}\\p(\vec{\omega})&=\underbrace{(2\pi)^{\frac{-d}{2}}\exp\bigg(\frac{-||\vec{\omega}||^2}{2}\bigg)}_{\text{Gaussian is symmetric around the origin}}\\&=\underbrace{(2\pi)^{\frac{-d}{2}}\exp\bigg(\frac{||\vec{\omega}||^2}{2}\bigg)}_{\text{just a Gaussian with 0 mean and unit variance}}\end{align}\)
Since the Gaussian is symmetric around the origin, we have shown that by drawing ω from the multivariate Gaussian with 0 mean and unit variance, the estimated value of the positive exponential feature map is equal to the following:
\(\small\begin{align}\mathbb{E}_{\vec{\omega}\sim \mathcal{N}(\vec{0}, \mathbf{I}_d)}\bigg[\exp(\vec{\omega}\cdot \mathbf{x})\exp(\vec{\omega}\cdot\mathbf{z})\bigg]&=\exp\bigg(\frac{||\mathbf{q+k}||^2}{2}\bigg)\int_{\mathbb{R}^d}(2\pi)^{\frac{-d}{2}}\underbrace{\exp\bigg(\frac{-||\vec{\omega}||^2}{2}\bigg)\exp\bigg(\frac{||\vec{\omega}||^2}{2}\bigg)}_{\exp(0)=1}\exp\bigg(-\frac{||\vec{\omega}-(\mathbf{q}+\mathbf{k})||^2}{2}\bigg)d\vec{\omega}\\&=\exp\bigg(\frac{||\mathbf{q+k}||^2}{2}\bigg)\underbrace{\int_{\mathbb{R}^d}(2\pi)^{\frac{-d}{2}}\exp\bigg(-\frac{||\vec{\omega}-(\mathbf{q}+\mathbf{k})||^2}{2}\bigg)d\vec{\omega}}_{\text{integral of normalized Gaussian = 1}}\\&=\exp\bigg(\frac{||\mathbf{q+k}||^2}{2}\bigg)\end{align}\)
Now that we have derived the expected value of the positive feature map, we need to figure out how to write the softmax function in terms of this expected value. First, we can observe that the term inside the exponential is a squared norm of the sum of two vectors. Since we already derived the squared norm of the difference between vectors, it is not hard to see that:10
By substituting this definition of the dot product into the softmax kernel, we get:
\(\begin{align}\text{SM}(\mathbf{q},\mathbf{k})&=\exp\left(\mathbf{q\cdot k}\right)\nonumber\\&=\exp\left(-\frac{||\mathbf{q}||^2}{2}-\frac{||\mathbf{k}||^2}{2}+\frac{||\mathbf{q}+\mathbf{k}||^2}{2}\right)\nonumber\\&=\exp\left(-\frac{||\mathbf{q}||^2}{2}\right)\exp\left(-\frac{||\mathbf{k}||^2}{2}\right)\underbrace{\exp\left(\frac{||\mathbf{q}+\mathbf{k}||^2}{2}\right)}_{\text{expected value of positive feature map!}}\end{align}\)
Observe that we can write the softmax kernel in terms of the expected value of the positive feature map:
\(\begin{align}\text{SM}(\mathbf{q},\mathbf{k})&=\exp\left(-\frac{||\mathbf{q}||^2}{2}\right)\exp\left(-\frac{||\mathbf{k}||^2}{2}\right)\underbrace{\mathbb{E}_{\vec{\omega}\sim \mathcal{N}(\vec{0}, \mathbf{I}_d)}\bigg[\exp(\vec{\omega}\cdot \mathbf{q})\exp(\vec{\omega}\cdot\mathbf{k})\bigg]}_{\text{expected value of positive feature map!}}\end{align}\)
We use the same approximation steps as before to derive our positive random feature estimator for r random feature vectors (denoted with a superscript + for positive):
\(\begin{align}\widehat{\text{SM}}^+_r(\mathbf{q},\mathbf{k})&=\exp\left(-\frac{||\mathbf{q}||^2}{2}\right)\exp\left(-\frac{||\mathbf{k}||^2}{2}\right)\underbrace{\frac{1}{r}\sum_{k=1}^r\exp(\vec{\omega}\cdot \mathbf{q})\exp(\vec{\omega}\cdot\mathbf{k})}_{\text{average over }r\text{ random feature projections}}\end{align}\)
Like the trigonometric estimator, we write the average as a dot product between two r-dimensional feature maps.
To derive the r-dimensional positive random feature map φ such that the dot product of the feature maps for vectors q and k approximates the softmax kernel, we can separate the terms into two equal expressions—one evaluated on q and the other on k.
Therefore, we have successfully derived a positive feature estimator for the softmax kernel that takes the dot product of the query and key vectors transformed by an r-dimensional feature map φ:
Given any d-dimensional vector x, the positive random feature map φ transforms the vector into an r-dimensional vector that encodes the data needed to approximate the softmax kernel after taking the dot product.
With similar steps as the trigonometric estimator, we can take out the constant terms and square them, then apply Bienaymé’s identity.
\(\begin{align}\small\text{MSE}\bigg(\widehat{\text{SM}}^+_r(\mathbf{q},\mathbf{k})\bigg)&=\text{Var}\left[\underbrace{\exp\left(\frac{-||\mathbf{q}||^2-||\mathbf{k}||^2}{2}\right)\frac{1}{r}}_{\text{constant (not dependent on }\omega)}\underbrace{\sum_{k=1}^r\exp\big(\vec{\omega}_k\cdot (\mathbf{q}+\mathbf{k})\big)}_{\text{random variable}}\right]\nonumber\\&=\underbrace{\frac{1}{r^2}\exp\left(-||\mathbf{q}||^2-||\mathbf{k}||^2\right)}_{\text{square constants}}\text{Var}\left[\sum_{k=1}^r\exp\big(\vec{\omega}_k\cdot (\mathbf{q}+\mathbf{k})\big)\right]\\&=\frac{1}{r^2}\exp\left(-||\mathbf{q}||^2-||\mathbf{k}||^2\right)\underbrace{r\text{Var}\bigg[\exp\big(\vec{\omega}\cdot (\mathbf{q}+\mathbf{k})\big)\bigg]}_{\text{variance of sum = sum of variances}}\\&=\underbrace{\frac{1}{r}\exp\left(-||\mathbf{q}||^2-||\mathbf{k}||^2\right)}_{\text{cancel }r}\text{Var}\bigg[\exp\big(\vec{\omega}\cdot (\mathbf{q}+\mathbf{k})\big)\bigg]\end{align}\)
Now, we will focus on expanding the variance term before substituting it back into our equation. Using the definition of variance as the expectation of the squared random variable subtracted by the squared of the expectation, we have:
Substituting this equality into our equation for variance, we get:
\(\small\begin{align}\text{Var}\bigg[\exp\big(\vec{\omega}\cdot (\mathbf{q}+\mathbf{k})\big)\bigg]&=\underbrace{\mathbb{E}\bigg[\exp\big(\vec{\omega}\cdot 2(\mathbf{q}+\mathbf{k})\big)\bigg]}_{\text{vectors are scaled by 2}}-\bigg(\mathbb{E}\bigg[\exp\big(\vec{\omega}\cdot (\mathbf{q}+\mathbf{k})\big)\bigg]\bigg)^2\\&=\underbrace{\exp\left(\frac{||2(\mathbf{q}+\mathbf{k})||^2}{2}\right)}_{\text{maintain scaled vectors}}-\left(\exp\left(\frac{||\mathbf{q}+\mathbf{k}||^2}{2}\right)\right)^2\\&=\underbrace{\exp\left(\frac{4||\mathbf{q}+\mathbf{k}||^2}{2}\right)-\exp\left(\frac{2||\mathbf{q}+\mathbf{k}||^2}{2}\right)}_{\text{cancel constants}}\\&=\underbrace{\exp\bigg(2||\mathbf{q}+\mathbf{k}||^2\bigg)-\exp\bigg(||\mathbf{q}+\mathbf{k}||^2\bigg)}_{\text{factor out }\exp\left(||\mathbf{q}+\mathbf{k}||^2\right)}\\&=\exp\left(||\mathbf{q}+\mathbf{k}||^2\right)\bigg(\exp\big(||\mathbf{q}+\mathbf{k}||^2\big)-1\bigg)\end{align}\)
Now, we can rewrite the MSE of the positive softmax estimator as:
Analyzing the equation above, we see that in the limit as the true value of the softmax functionapproaches 0, the MSE of the positive random feature estimator is equal to 0.
We can also observe that the MSE is inversely proportional to the number of random feature vectors r, which confirms our intuition that as the number of random features increases, so does the precision of our approximation.
Therefore, we have proven that the positive random feature estimator is exponentially more accurate than the trigonometric random feature estimator for approximating the softmax function for small outputs.
But what about the limit as the softmax function approaches infinity?
Although it might seem as if the MSE will approach infinity as the softmax function grows large; however, in the next sections, we will discuss the normalization steps used to ensure that the softmax value is bounded between 0 and 1 (just like in normal attention) and the features of query and key vectors are normalized based on their dimension d.
We can visualize this in the graph below which shows the ratio between the MSE for the trigonometric random feature estimator divided by the MSE for the positive random feature estimator, which increases to infinity as the angle between the query and key vectors approaches 90 degrees (small dot product; low attention scores). In the top-right panel, we see that the MSE for the positive random feature estimator remains low even as the angles approach zero, with only a slight increase.
Graph of the ratio of the theoretical mean-squared error (MSE) for the trigonometric estimator divided by the MSE for the positive estimator as a function of the angle between the input vectors (radians) and the vector length. When the angle between the query and key vectors is close to orthogonal (small dot product; low attention scores), the MSE for the trigonometric estimator is arbitrarily more inaccurate than the positive estimator. The top-right panel shows the MSE for the trig and positive estimators separately. (Source)
Experimental results also showed that the measured MSE of the trigonometric estimator is orders of magnitude higher than the MSE of the positive estimator even as the number of random feature vectors (r) increases. The positive feature estimator maintained a low MSE even with only 25 random feature projections when the input vectors had dimension d = 16.
MSE of softmax approximation with the trigonometric vs the positive random feature estimators as a function of the number of random feature vectors. This was measured on an input sequence of length N = 4096 and an embedding dimension of d = 16. (Source)
This result is quite incredible. With just a simple tweak in the type of function used in the estimator, we are able to lower MSE by full orders of magnitude.
Since there is an abundance of low attention scores across sequences at variable positions in the query MSA that are crucial for capturing the contextual information across homologs, positive random feature estimators are necessary to precisely approximate long-range dependencies across the MSA.
Now that we understand the rationale behind the ‘+’ of FAVOR+, let’s dive into the ‘O’ for orthogonal random features.
Orthogonal Random Features
By orthogonalizing the random feature vectors sampled from the multivariate Gaussian, we minimize the number of random feature projections, r, needed to accurately approximate the softmax function. This is done with QR decomposition using the Gram-Schmidt process.
In the last section, we derived the MSE of the positive random feature estimator to be:
From this equation, we can observe that the MSE decreases linearly as the number of random features increases. However, more random feature vectors translate to larger computational time and space complexity.
This raises the question: How can we minimize the number of random feature vectors r without sacrificing the accuracy of the estimator? In other words: For a given number of random features r, how do we sample an optimal subset of vectors that minimizes the mean-squared error?
This is where orthogonal random features come in.
A set Ω of r mutually orthogonal vectors has several properties that make it the best representative subset for the r-dimensional vector space (denoted V).
Orthogonal vectors are linearly independent, meaning that no vector in the set can be written as a linear combination (weighted sum) of the other vectors in the set.
This implies that each orthogonal vector spans a unique subspace of the full feature space that is not spanned by any of the other vectors.
The set Ω of r orthogonal vectors is a minimally spanning subset of V. This means that if we remove any vector in Ω, the set Ω is no longer a spanning set of the entire feature space.
The set Ω is a maximal linearly independent subset of V. This means that if we add a vector to Ω, the set will become linearly dependent. In other words, any additional r-dimensional vector can be written as a linear combination of the vectors in Ω.
Therefore, expanding the set Ω will not contribute significantly to the accuracy of the approximation.
In fact, any estimator (positive or trigonometric) for the softmax kernel (and other kernels including the RBF kernel) using orthogonal random features has lower MSE than the same estimator using independently and identically drawn random features. The decrease in MSE is larger as the number of random features rincreasesand as the true value of the softmax function increases.
\(\underbrace{\text{MSE}\bigg(\widehat{\text{SM}}^{\text{ort}}_r(\mathbf{q, k})\bigg)}_{\text{MSE for orthogonal random features}}\leq \underbrace{\text{MSE}\bigg(\widehat{\text{SM}}^{\text{iid}}_r(\mathbf{q, k})\bigg)}_{\text{MSE for i.i.d. random features }}-\underbrace{\left(1-\frac{1}{r}\right)}_{\text{increases as }r\text{ increases
}}\underbrace{\left(\frac{2}{d+2}\right)}_{\text{decreases as }d\text{ increases}}\underbrace{\bigg(\text{SM}(\mathbf{q, k})-a_0\bigg)^2}_{\text{increases with true SM}}\)
As the input feature dimension d increases, the difference between the orthogonal and iid estimators shrinks; however, for any d > 0, the orthogonal feature estimator performs better than the iid feature estimator, meaning that orthogonal random features are universally superior in performance.
Since it is quite intuitive that maximizing the feature space spanned by the random feature vectors decreases the number of random vectors needed, I will omit the lengthy proof which can be found in Appendix F.4.1 of the Performer paper.
Now, let’s discuss the optimal range for the number of random features r.
In Theorem 4, Choromanski et al. proved that the number of random projections r needed to approximate any attention matrix A up to any precision is only dependent on the embedding dimension d and not on the dimensions of the attention matrix itself, N.
For an error ϵ > 0, we define the precision as the maximum difference between an element in the estimated attention matrix A hat, and true attention matrix A which is equivalent to the infinite-norm of the difference between the two matrices.
\(\underbrace{||\hat{\mathbf{A}}-\mathbf{A}||_{\infty}}_{\text{max difference predicted and true scores}}\leq \epsilon\)
The lower bound for the number of random features r needed to achieve the error ϵ is given by11:
d is the dimension of the input vectors. If all other variables are held constant, then the optimal number of random features is on the order of dlog(d) (larger than d but smaller than d²).
R is the maximum L2 norm of the input query and key vectors. As R increases, the number of random features for ϵ error increases logarithmically. Since R increases as the subspace covered by the queries and keys increases, this means that for long enough sequences that cover a larger vector space, a fixed r may result in increased error.
q~ and k~ are the maximum values of the exponential Gaussian centered at 0 evaluated for q and k, meaning they are bounded by (0, 1]. This value is used to scale the random feature map of the RBF kernel to obtain the softmax kernel. q~ and k~ decrease as the distance of the query and key vectors from the origin decreases, so the number of random features needed decreases as the queries and keys move closer to the origin.
\(\begin{align}q^*=\underbrace{\max_{\mathbf{q}}\left(\exp\left(\frac{-||\mathbf{q}||^2}{2}\right)\right)}_{\text{maximum term that scales the RBF feature map for all }\mathbf{q}}\\k^*=\underbrace{\max_{\mathbf{k}}\left(\exp\left(\frac{-||\mathbf{k}||^2}{2}\right)\right)}_{\text{maximum term that scales the RBF feature map for all }\mathbf{k}}\end{align}\)
Since d is the only variable that varies largely across applications, this result demonstrates that defining r on the order of dlog(d) allows us to approximate A up to any precision defined by ϵ in only O(Nd²log(d)) time.
\(\underbrace{O(Nd^2\log(d))}_{\text{time complexity of orthogonal random feature estimator}}\)
Notice that since d does not grow with batch size or sequence dimension, this reduces the time complexity to be linearly dependent on N rather than quadratically dependent on N like in regular column-wise attention.
Now that we understand the purpose of orthogonal random features and the number of random features needed to approximate the softmax kernel, let’s break down the theory and implementation for computing a set of r uniformly distributed orthogonal feature vectors.
Since sampling from a multivariate Gaussian with covariance 0 is the same as independently sampling each entry from a 1-dimensional Gaussian, we can easily generate a set of d-dimensional random feature vectors as a d x r matrix M where each entry is an independently and identically sampled random feature from the single variable Gaussian distribution with a mean of 0 and unit variance.
Since the maximum number of mutually orthogonal vectors in d-dimensional space is dand r is on the order of dlog(d), we first split Minto several d x d square matrices and orthogonalize each chunk separately. This will give us multiple sets of d mutually orthogonal vectors that, when put together form a set of r “almost” orthogonal vectors.
Using the QR decomposition, we convert each d x dsquare matrix M into an orthonormal matrixQ, where each column is mutually orthogonal with unit length. The resulting matrix Q is related to M by multiplication with an upper-triangular matrix R such that the following identity holds:
Although several algorithms can be used for QR decomposition, we will break down the most common and intuitive method: the Gram-Schmidt process.
\(\begin{align}\underbrace{\begin{bmatrix}|&|&&|\\\mathbf{m}_1&\mathbf{m}_2&\dots &\mathbf{m}_d \\|&|&&|\\\end{bmatrix}}_{\text{random feature matrix}}\xrightarrow{\text{Gram-Schmidt process}}\underbrace{\begin{bmatrix}|&|&&|\\\vec{\omega}_1&\vec{\omega}_2&\dots &\vec{\omega}_d \\|&|&&|\\\end{bmatrix}}_{\text{columns are mutually orthogonal and unit length}}\end{align}\)
The columns of the resulting matrix satisfy the following properties:
Before we get started, let’s define some key properties of vectors.
The vectorprojection of a onto the line spanned by the vector b can be computed by multiplying the dot product by the unit vector in the direction of vector b.
\(\text{proj}_{\mathbf{b}}\mathbf{a}=\underbrace{\underbrace{(\mathbf{a}\cdot \mathbf{b})}_{\text{length}}\underbrace{\frac{\mathbf{b}}{||\mathbf{b}||}}_{\text{unit vector of }\mathbf{b}}}_{\in \mathbb{R}^d}\tag{$\mathbf{a},\mathbf{b}\in {\mathbb{R}^d}$}\)
If b is already a unit vector (||b|| = 1), then we do not have to normalize by the length and can directly multiply the dot product (a • b) with b.
The Gram-Schmit process orthogonalizes one column at a time such that it is orthogonal to all the columns to its left.
Since the first column has no columns to its left, we simply divide each element by its length (L2 norm) to get a unit vector pointing in the same direction.
To make the second column m_2 orthogonal to the first column ω_1, we take the vectorprojection of m_2 onto the line spanned by ω_1 and subtract it from m_2. This gives us a vector u_2 (not necessarily of unit length) orthogonal to ω_1.
The intuition behind why u_2 is orthogonal to ω_1 is simple. Since the vector projection of m_2 onto ω_1 forms a right angle with the line spanned by ω_1, we know that m_2 can be written as the vector sum of the projection and some orthogonal vector.
\(\mathbf{m}_2=\underbrace{\text{proj}_{\vec{\omega}_1}\mathbf{m}_2}_{\mathbf{m}_2\text{ in the direction of } \vec{\omega}_1}+\underbrace{\mathbf{u}_2}_{\text{orthogonal to } \vec{\omega}_1}\)
So, to solve for u_2, we simply rearrange the equation to get:
For the kth column, we need to make it orthogonal to all k-1 columns to its left. To do this, we generalize Step 2 by projecting of the m_k on each of the first k-1 orthonormal column vectors from the matrix Q. Then, we subtract all k-1 projections from m_k to get the component of m_i that cannot be constructed as a linear combination of the k-1 orthonormal vectors, meaning it is mutually orthogonal to all k-1 vectors.
We repeat Steps 4 and 5 for all d columns in M such that the last column is mutually orthogonal to all d-1 columns.
Now that we understand how the QR decomposition works, we can implement it with the built-in PyTorchtorch.linalg.qr function. Let’s define a function orthogonal_matrix_chunk that given a dimension d, returns a d x d orthonormal matrix M with mutually orthogonal columns.
We start by sampling a d x d square matrix of random features independently and identically from a Gaussian distribution with a mean of 0 and unit variance using the torch.randnfunction.
Next, we call torch.linalg.qr on the d x d matrix of iid random features which applies QR decomposition to output a d x d orthonormal matrix Q and d x d upper-triangular matrix R.
Since we want the orthogonal vectors to be along the rows of the final r x d matrix, the function orthogonal_matrix_chunk returns the transpose of Q.
We then define the function gaussian_orthogonal_random_matrix that concatenates each d x d orthogonal block into a single r x d matrix of “almost” orthogonal vectors and rescales each vector such that their lengths (L2 norms) represent a uniformly sampled set of vectors.
Since QR decomposition converts all the vectors to unit length, we need to rescale each vector to match a randomly sampled vector from the multivariate Gaussian. This ensures that the lengths of the random vectors are distributed similarly to the original random feature vectors. To do this, we sample another r x d matrix of random features W.
Then, we take the L2 norm of each row vector w to get an r-dimensional vector of lengths that will be used to scale each row of the random orthogonal matrix M.
By arranging the norms along the diagonal of an r x r matrix and taking the matrix product with the matrix of random orthogonal vectors, we get the final r x d matrix of random Gaussian orthogonal vectors, each scaled to match the length of another randomly generated vector.
Now that we have a set of r-dimensional orthogonal features, we can finally implement the function that returns the r-dimensional feature maps given an array of query or key vectors.
We define a function called softmax_kernel that takes the array of either query or key embeddings with shape (B*L, H, N, d) for sequence-wise axial attention and a r x d matrix of orthogonal random feature vectors and returns the (B*L, H, N, r) array of transformed query* or key* vectors.
In general, this function takes a d-dimensional vector and projects it into a r-dimensional positive random feature map with the following equation:
Until now, we have omitted the normalization by √d applied after the dot product in the regular attention mechanism. To incorporate the normalization step in our feature map, we have:
\(\begin{align}\text{SM}(\mathbf{q},\mathbf{k})&=\exp\left(\frac{\mathbf{q\cdot k}}{\sqrt{d}}\right)\nonumber\\&=\exp\left(-\frac{||\mathbf{q}||^2}{2\sqrt{d}}\right)\exp\left(-\frac{||\mathbf{k}||^2}{2\sqrt{d}}\right)\underbrace{\exp\left(\frac{||\mathbf{q}+\mathbf{k}||^2}{2\sqrt{d}}\right)}_{\text{need to separate normalization for }\mathbf{q}\text{ and }\mathbf{k}}\end{align}\)
Since the last exponential term is approximated using the random feature map, we need to scale the query and key independently with a normalization term. Therefore, we rewrite the normalized expression as follows:
This allows us to approximate the normalized exponential term by scaling the query and key vectors down by the fourth root of d, and we can write the final normalized positive random feature map as:
Now, we can walk through the implementation of the softmax_kernel function which computes the terms inside exponentials before putting it all together using matrix multiplication. The number below each term corresponds to a step below.
First, we extract the relevant dimensions and compute a normalizer equal to the fourth root of d which will scale each input vector before taking the dot product with the random feature vectors.
Next, we compute the ratio 1/√r which takes the average across all random feature projections.
Since we are projecting all embeddings across each sequence in the batch and each attention head, we repeat the r x d matrix of orthogonal random features along the attention headdimension and the batch dimension to get an array with shape (B*L, H, r, d).
We can now take the matrix product of the N x d matrix of the input feature matrix and the transposed d x r matrix of orthogonal random feature vectors. This is equivalent to taking the dot product between every pair of input embedding and random vector, producing an N x r matrix of transformed embeddings along each row.
In the code implementation, we use torch.einsum which takes the (B*L, H, N, d)normalized data array with the (B*L, H, r, d) random feature array and computes the matrix product along the last two dimensions to output an array with shape (B*L, H, N, r).
Now we will compute the exponential term that scales each entry of the random feature map. We start by taking the squared L2 norm of each d-dimensional vector in data by squaring the entries of the N x d input matrix and summing over each row.
\(\begin{bmatrix}—&\mathbf{x}_1^2&—\\—&\mathbf{x}_2^2&—\\&\vdots\\—&\mathbf{x}_N^2&—\end{bmatrix}\xrightarrow{\text{sum each row}}\begin{bmatrix}||\mathbf{x}_1||^2\\||\mathbf{x}_2||^2\\\vdots\\||\mathbf{x}_N||^2\end{bmatrix}\)
Next, we scale down each squared norm by 2√d and add a singleton dimension to the last dimension of the array to get an array with shape (B*L, H, N, 1) which will be broadcasted to scale each row of the array of random feature projections with shape (B*L, H, N, d).
\(\begin{bmatrix}||\mathbf{x}_1||^2\\||\mathbf{x}_2||^2\\\vdots\\||\mathbf{x}_N||^2\end{bmatrix}\xrightarrow{\text{scale down and broadcast}}\underbrace{\begin{bmatrix}\frac{||\mathbf{x}_1||^2}{2\sqrt{d}}&\frac{||\mathbf{x}_1||^2}{2\sqrt{d}}&\dots&\frac{||\mathbf{x}_1||^2}{2\sqrt{d}}\\\frac{||\mathbf{x}_2||^2}{2\sqrt{d}}&\frac{||\mathbf{x}_2||^2}{2\sqrt{d}}&\dots &\frac{||\mathbf{x}_2||^2}{2\sqrt{d}}\\\vdots&\vdots&\ddots&\vdots\\\frac{||\mathbf{x}_N||^2}{2\sqrt{d}}&\frac{||\mathbf{x}_N||^2}{2\sqrt{d}}&\dots&\frac{||\mathbf{x}_N||^2}{2\sqrt{d}}\end{bmatrix}}_{N \times r}\)
Since the softmax function normalizes all the dot products generated from a single query embedding and the set of key embeddings to be a value between 0 and 1, we want the transformed query* and key* vectors to have entries in the range [0, 1] to prevent the exponential function from growing too large.
Exponential function e^x. Notice that negative values are within the bounds [0, 1]. (Made with Desmos)
Since the exponential function evaluated at all negative values is between (0, 1] we can convert all the entries of the random feature map to be negative by subtracting the maximum entry across the vector.
Since each query vector computes a new set of attention scores, we subtract the maximum element within its row so that after applying the exponential, the maximum element of each row is 1.
\(\phi(\mathbf{q})=\frac{1}{\sqrt{r}}\left(\exp\left(-\frac{||\mathbf{q}||^2}{2}\right)\exp\left(\begin{bmatrix}
\frac{\vec{\omega}_1\cdot\mathbf{q}}{\sqrt[4]{d}}\\
\\
\frac{\vec{\omega}_2\cdot\mathbf{q}}{\sqrt[4]{d}}\\
\\
\vdots\\
\\
\frac{\vec{\omega}_r\cdot\mathbf{q}}{\sqrt[4]{d}}
\end{bmatrix}-\underbrace{\max_k\left(\frac{\vec{\omega}_k\cdot\mathbf{q}}{\sqrt[4]{d}}\right)}_{\text{max element for single query map}}\right)+\epsilon\right)\)
Since each set of attention scores is computed using all N key vectors, we subtract the maximum element across the random feature maps for all the key vectors so that after applying the exponential, the maximum element across the entire feature map is 1.
\(\phi(\mathbf{k}_n)=\frac{1}{\sqrt{r}}\left(\exp\left(-\frac{||\mathbf{k}_n||^2}{2}\right)\exp\left(\begin{bmatrix}
\frac{\vec{\omega}_1\cdot\mathbf{k}_n}{\sqrt[4]{d}}\\
\\
\frac{\vec{\omega}_2\cdot\mathbf{k}_n}{\sqrt[4]{d}}\\
\\
\vdots\\
\\
\frac{\vec{\omega}_r\cdot\mathbf{k}_n}{\sqrt[4]{d}}
\end{bmatrix}-\underbrace{\max_{k,n}\left(\frac{\vec{\omega}_k\cdot\mathbf{k}_n}{\sqrt[4]{d}}\right)}_{\text{max element across all key maps}}\right)+\epsilon\right)\)
Finally, the function returns the normalized random feature map.
Multi-Head Fast Attention
Here, we will implement the multi-head fast attention mechanism with the help of matrix multiplication and the einsum function in PyTorch.
To implicitly compute the updated MSA embeddings from the (B*L, H, N, r)transformed query array, the (B*L, H, N, r) transformed key array, and the (B*L, H, N, d) value embedding array, we define a function called linear attention which executes the attention mechanism with only matrix multiplication following the equation:
Let’s write the above equation with vector notation to show how we incorporate the normalization across the set of attention scores like the regular softmax function.
Comparison between sequence-wise multi-head attention with softmax random feature maps and regular attention with the softmax function. (Source: Alchemy Bio)
For sequence-wise attention across the column corresponding to position i, we can write the regular softmax attention mechanism for position i in sequence n as follows:
We’ve accounted for the normalization by √d in the previous section, so we can simply substitute our random feature approximation into the equation above.
The query vector is neither dependent on m in the numerator nor m’ in the denominator, so we can take it out from the summation. Note, however, that in matrix arithmetic, we cannot simply cancel out the query* vector in the numerator and denominator.
We now observe that the product of a column vector (transposed key in the numerator) and a row vector (value) is equivalent to the outer product of the two vectors, denoted by ⊗.
With this definition, we can expand the sum of the outer products between the key* and value vectors into the matrix product of the d x N transposed key* matrix and the N x d value matrix.
Now that we understand the underlying matrix multiplication steps, converting this into code is less intimidating. Here, I will provide the implementation for each operation using torch.einsumand give some alternative implementations with torch.matmulfor clarity. To refresh on the common operations using einsum, see the preface section.
First, we compute the sum of the aggregated key* column vector in the denominator of the normalization term by taking the sum over the sequence dimension N of the (B*L, H, N, d) key array.
Next, we compute the normalization term by taking the inverse of the matrix product of the N x d query* matrix and the d-dimensional aggregated key* column vector which gives an N-dimensional vector where each entry is the normalization term for a single sequence that sequence n is attending to.
For implementation with matmul, we add a singleton dimension to convert the summed key* array to shape (B*L, H, d, 1) before taking the matrix product with the (B*L, H, N, d) query* array.
Alternatively, we can implement this with einsum without adding a singleton dimension.
Now, we take the matrix product of the d x N transposed key* matrix and the N x d value matrix to get the intermediate d x d matrix.
We can implement this by calling matmul on the (B*L, H, d, N) transposed key* array and (B*L, H, N, d) value array.
With einsum, we set N as the dimension being summed over and the last dimensionsof the key* array with shape (B*L, H, N, d) and the value array with shape (B*L, H, N, d) as the output dimensions to compute the outer product intermediate array with shape (B*L, H, d, d).
After deleting the key* and value arrays to reduce memory, we normalize the query* vectors by scaling each row of the N x d query* matrix by a single element of the N-dimensional normalization vector. This is equivalent to repeating the N-dimensional normalization vector d times along each column of an N x d matrix and taking the element-wise product with the N x d query* array.
We can implement this with einsum without needing to repeat the columns with the code below, where for each index in the leading dimensions (…), the nth element of D_inv multiplies the corresponding nth row of q.
Finally, we compute the updated contextual MSA embeddings by taking the matrix product of the N x d normalized query* matrix and the d x d intermediate matrix.
We implement this using einsum with the definition of matrix multiplication described in the preface.
The full linear_attention function implemented with torch.einsum is given below:
Now we can define the module called FastAttention that initializes the random feature projection array with shape (r, d), where r is set by default to r = dlog(d) and stores them as buffers in the module state. The module also defines a function to re-sample the random feature vectors.
The forward function takes the projected query, key, and value arrays with shape (B*L, H, N, d), computes the transformed query* and key* random feature maps, and calls the linear_attention function to return the final contextual embedding array with shape (B*L, H, N, d).
The FastAttention module is encapsulated in the SelfAttention module which projects the input embeddings into q, k, and v vectors, splits the projections into multiple heads, calls the FastAttention module, and combines the embeddings across heads using a linear transformation. Since we already described a similar module in the section on Multi-head Axial Attention, I have omitted the full module here.
Axial Encoder
The axial encoder module combines the attention mechanisms that we have described into a single encoder network that applies residue-wise attention and sequence-wise attention followed by a feed-forward layer, with residual connections and normalization layers in between.
The AxialEncoderLayer module has three overall mechanisms: residue-wise attention, sequence-wise attention, and a feed-forward layer. We apply a pre-normalization layer before and a dropout layer after each mechanism.
Axial encoder module for MSA to MSA updates. The full array of N x Ld_msa-dimensional embeddings are fed into a soft-tied residue-wise attention module, a fast sequence-wise attention module, and a feed-forward layer to generate the updated contextual embeddings. (Source: Alchemy Bio)
In the constructor, we initialize the modules needed for each mechanism:
First, we normalize the input embedding array and feed it into the residue-wise attention layer. For tied and soft-tied attention, we want to share attention scores only across the template sequences aligned with the same query sequence so we keep batch and sequence dimensions isolated in the input array with shape (B, N, L, d_msa).
In contrast, fast linearized attention with the Performer architecture and regular multi-head self-attention both compute a new set of attention scores for all N sequences in the MSA, so we can concatenate the batch and sequence dimensions into a single dimension with shape (B*N, L, d_msa) to parallelize the attention mechanism across all sequences in the array.
Since the query, key, and value embeddings are all generated by transforming the input MSA embeddings, all three inputs to the residue- and column-wise attention modules are the same array of MSA embeddings.
When we discuss direct multi-head attention and cross multi-head attention later to integrate features across different tracks of the three-track architecture, the input embeddings used to generate the queries will differ from the keys and values.
Finally, we apply dropout to the output embedding array and add the input MSA embeddings as a residual connection.
Residual connections in deep learning is a technique used to prevent vanishing gradients by adding or concatenating input data to the transformed output data between layers which allows the gradient to skip past gradient-diminishing layer operations during backpropagation (quote from my previous article).
For sequence-wise attention, we permute the input embedding array with shape (B, N, L, d_msa) to shape (B, L, N, d_msa). For tied multihead attention, we leave the batch and residue dimension separated, but for untied attention, we concatenate the batch and residue dimensions into shape (B*L, N, d_model) to parallelize the attention mechanism across all columns and all MSAs in the batch.
Finally, we apply a feed-forward layer with a hidden dimension of d_ff which projects the MSA embeddings to higher dimensional d_ff-dimensional feature space, applies a nonlinear ReLU activation function, and projects each embedding back down to the original d_msa-dimensional feature space.
We call the AxialEncoder module inside the MSA2MSA class which takes the initial MSA embedding array with the same embedding across each amino acid type, a positional encoding, and a query encoding and captures contextual features across multiple representation subspaces of the protein.
With that, we are done with the MSA embedding module. As you may know, there is no shortage of new attention architectures being designed and iterated on, and many of them have the potential to enhance the expressiveness of MSA embeddings. For instance, Alphafold2’s Evoformer leverages Triangular Self-Attention, and ESMFold uses Local Attention and Locality Sensitive Hashing (LSH) Attention. Still, these mechanisms build on the same fundamental ideas as the algorithms described here.
Looking Foward
That’s it for Part 1 of 2 of my Complete Guide to Protein Structure Prediction with RoseTTAFold. I have split this guide into two parts to prevent overwhelming you with the full architecture at once.
In Part 2, I will be covering the rest of the 3-track architecture including the 2D track (template embeddings, pair features), the 3D track (graph transformer, SE(3)-transformer) as well as the mechanisms used to pass information across tracks (direct attention, masked attention).
To get notified when Part 2 is released and follow along in my journey through more deep dives into bio-ML concepts, consider subscribing and supporting me further by becoming a paid subscriber ($12 a month).
For a sneak peek, here is a diagram of the full RoseTTAFold architecture:
Full three-track protein prediction architecture. The 1D track updates the MSA embeddings, the 2D track updates the pair features, and the 3D track updates a 3D geometric graph representation. (Source: Alchemy Bio)
Thank you for reading! This post is part 1 of a 2-part series written for those looking to fully grasp the underlying mechanisms of the protein structure prediction models. If you have any suggestions, please feel free to reach out via LinkedIn. Listed below are the journal articles I referenced while writing this article:
Minkyung Baek et al., “Accurate Prediction of Protein Structures and Interactions Using a Three-Track Neural Network,” Science 373, no. 6557 (August 20, 2021): 871–76, https://doi.org/10.1126/science.abj8754.
Minkyung Baek et al., “Efficient and Accurate Prediction of Protein Structure Using RoseTTAFold2” (bioRxiv, May 25, 2023), https://doi.org/10.1101/2023.05.24.542179.
Michael Remmert et al., “HHblits: Lightning-Fast Iterative Protein Sequence Searching by HMM-HMM Alignment,” Nature Methods 9, no. 2 (February 2012): 173–75, https://doi.org/10.1038/nmeth.1818.
Martin Steinegger et al., “HH-Suite3 for Fast Remote Homology Detection and Deep Protein Annotation,” BMC Bioinformatics 20, no. 1 (September 14, 2019): 473, https://doi.org/10.1186/s12859-019-3019-7.
Stephen F. Altschul et al., “Gapped BLAST and PSI-BLAST: A New Generation of Protein Database Search Programs,” Nucleic Acids Research 25, no. 17 (September 1, 1997): 3389–3402, https://doi.org/10.1093/nar/25.17.3389.
Gaston H. Gonnet, Mark A. Cohen, and Steven A. Benner, “Exhaustive Matching of the Entire Protein Sequence Database,” Science 256, no. 5062 (June 5, 1992): 1443–45, https://doi.org/10.1126/science.1604319.
S. Henikoff and J. G. Henikoff, “Amino Acid Substitution Matrices from Protein Blocks,” Proceedings of the National Academy of Sciences of the United States of America 89, no. 22 (November 15, 1992): 10915–19, https://doi.org/10.1073/pnas.89.22.10915.
Jonathan Ho et al., “Axial Attention in Multidimensional Transformers” (arXiv, December 20, 2019), http://arxiv.org/abs/1912.12180.
John Jumper et al., “Highly Accurate Protein Structure Prediction with AlphaFold,” Nature 596, no. 7873 (August 2021): 583–89, https://doi.org/10.1038/s41586-021-03819-2.
Joseph L. Watson et al., “De Novo Design of Protein Structure and Function with RFdiffusion,” Nature 620, no. 7976 (August 2023): 1089–1100, https://doi.org/10.1038/s41586-023-06415-8.
Alchemy Bio is a blog where I share transformational ideas in computational biology, synbio, and biotech. If you don’t want to miss upcoming blogs, you should consider subscribing for free to have them delivered to your inbox:
Big-O notation upper bounds a variable or run-time. If a variable or run-time is in O(f(d)), this implies that it is less than or equal to some constant c multiplied by f(d).
A hyperplane in linear machine learning models is a d-dimensional plane defined by the normal vector to the plane w. This hyperplane is found by learning the separation of data points in a training dataset and fitting the plane to best separate the data for binary classification. For each data point x, the model classifies x by taking its dot product with normal to the hyperplane w to get the distance (or margin) of the point to the hyperplane and the side of the plane that it is on (sign of the dot product). An understanding of linear models is not essential for understanding Performers, but if you would like to learn more, see these lecture notes.
The full derivation of the dimension of the polynomial kernel feature map can be found here. But, the biggest takeaway here is that the dimension grows with the degree of the polynomial and the dimension of the input features.
Some resources that I found extremely helpful for conceptualizing the Fourier transform include these lecture notes by David Morin and this video lecture by Steve Brunton.
The proof for the variance of a random variable that is scaled by a constant is given as:
\(\small\begin{align}\text{Var}[cX]&=\mathbb{E}[c^2X^2]-\big(\mathbb{E}[cX]\big)^2\\&=\underbrace{c^2\mathbb{E}[X^2]-\big(c\cdot \mathbb{E}[X]\big)^2}_{\text{expected value of constant is itself}}\\&=c^2\mathbb{E}[X^2]-c^2\big(\mathbb{E}[X]\big)^2\\&=c^2\underbrace{\bigg(\mathbb{E}[X^2]-\big(\mathbb{E}[X]\big)^2\bigg)}_{\text{definition of variance of }X}\\&=c^2\text{Var}[X]\end{align}\)








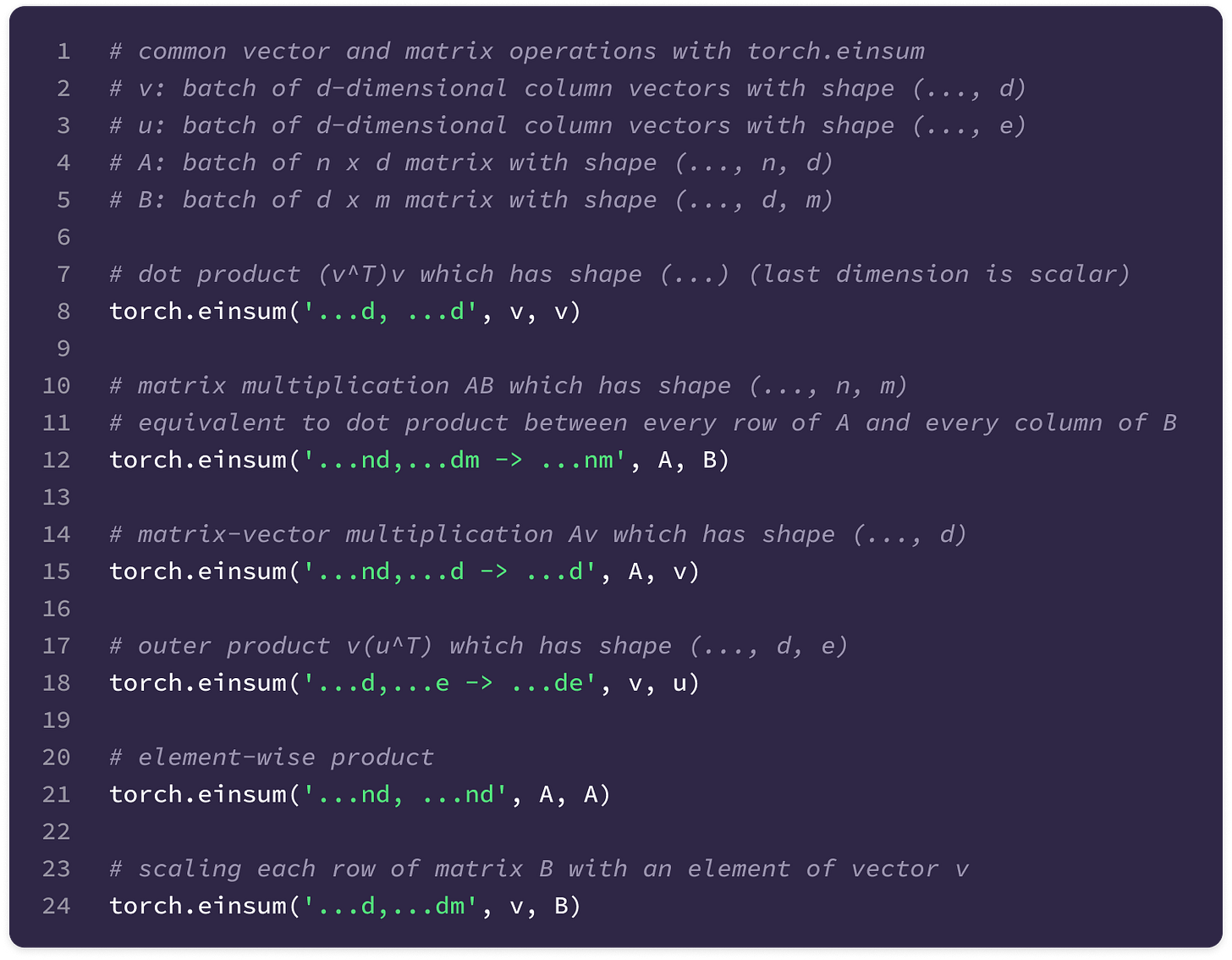

















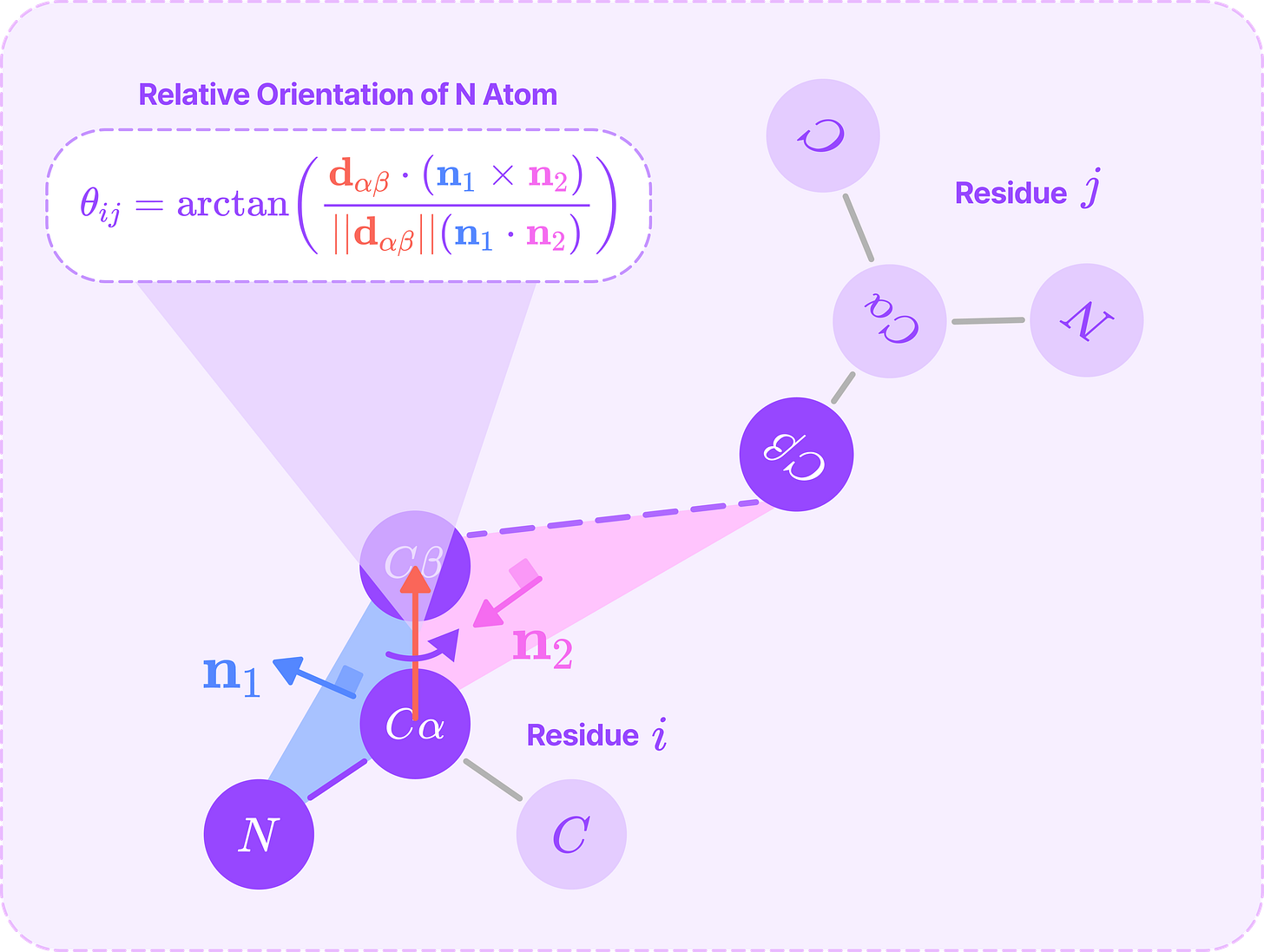




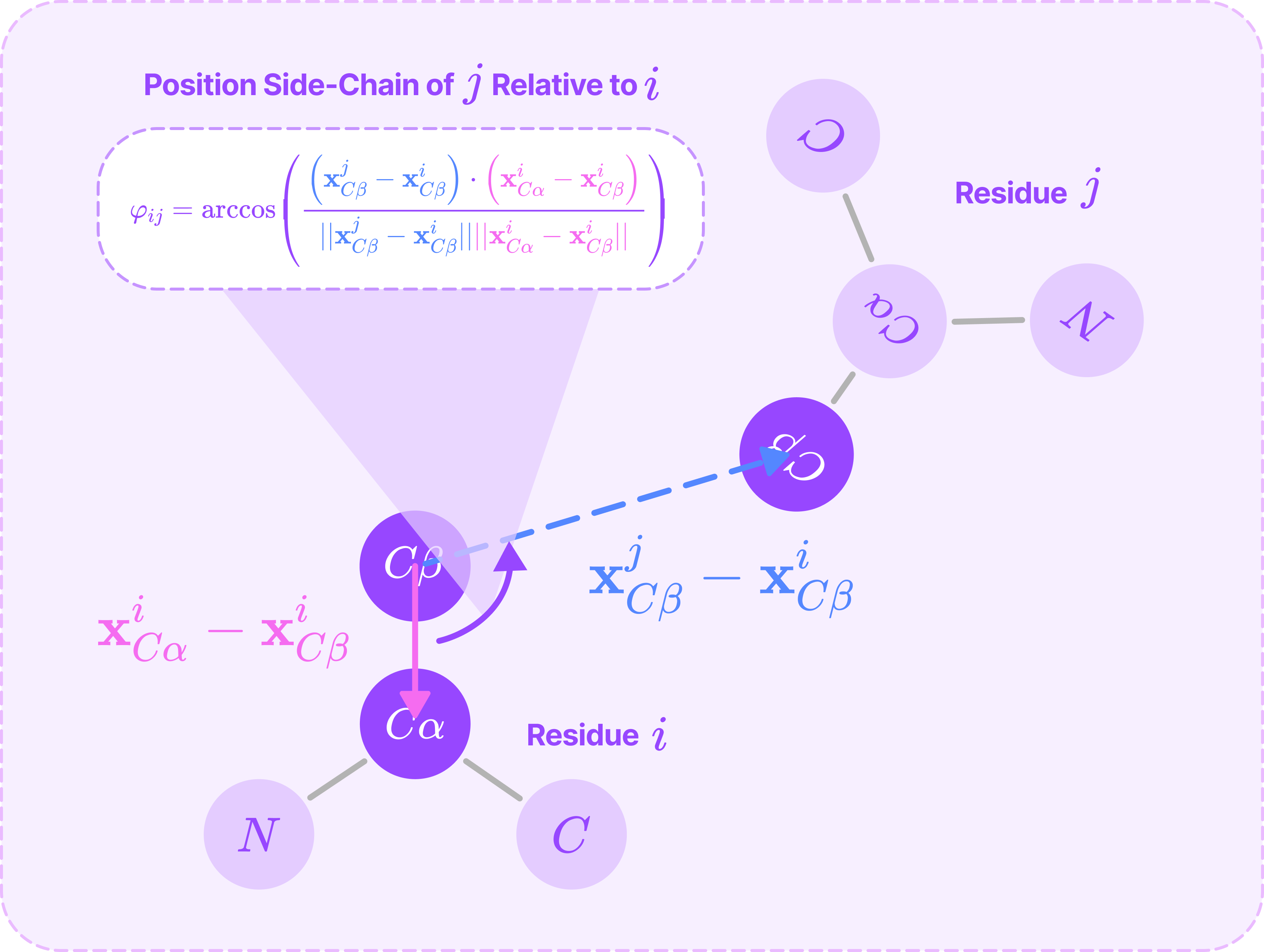




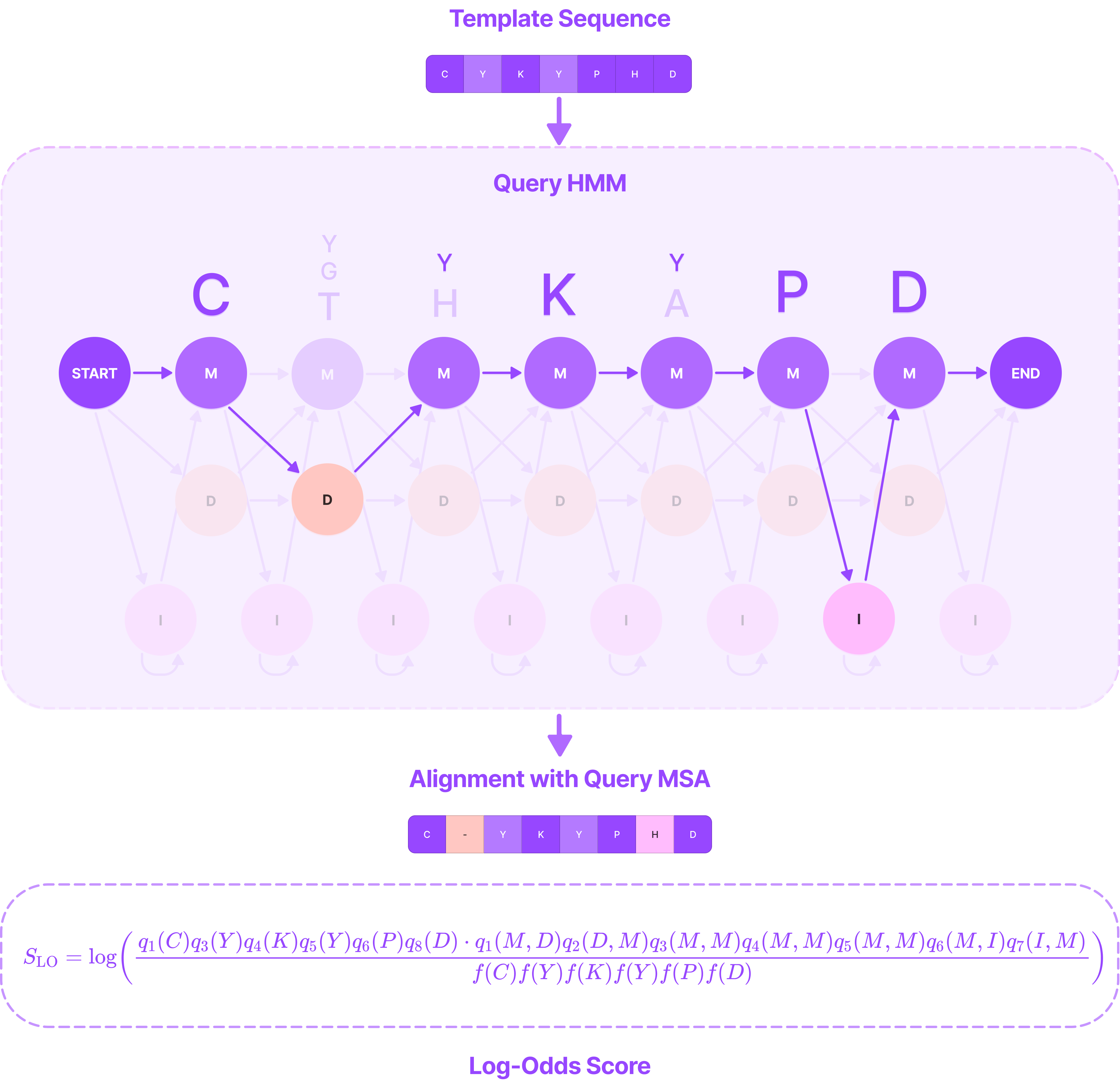
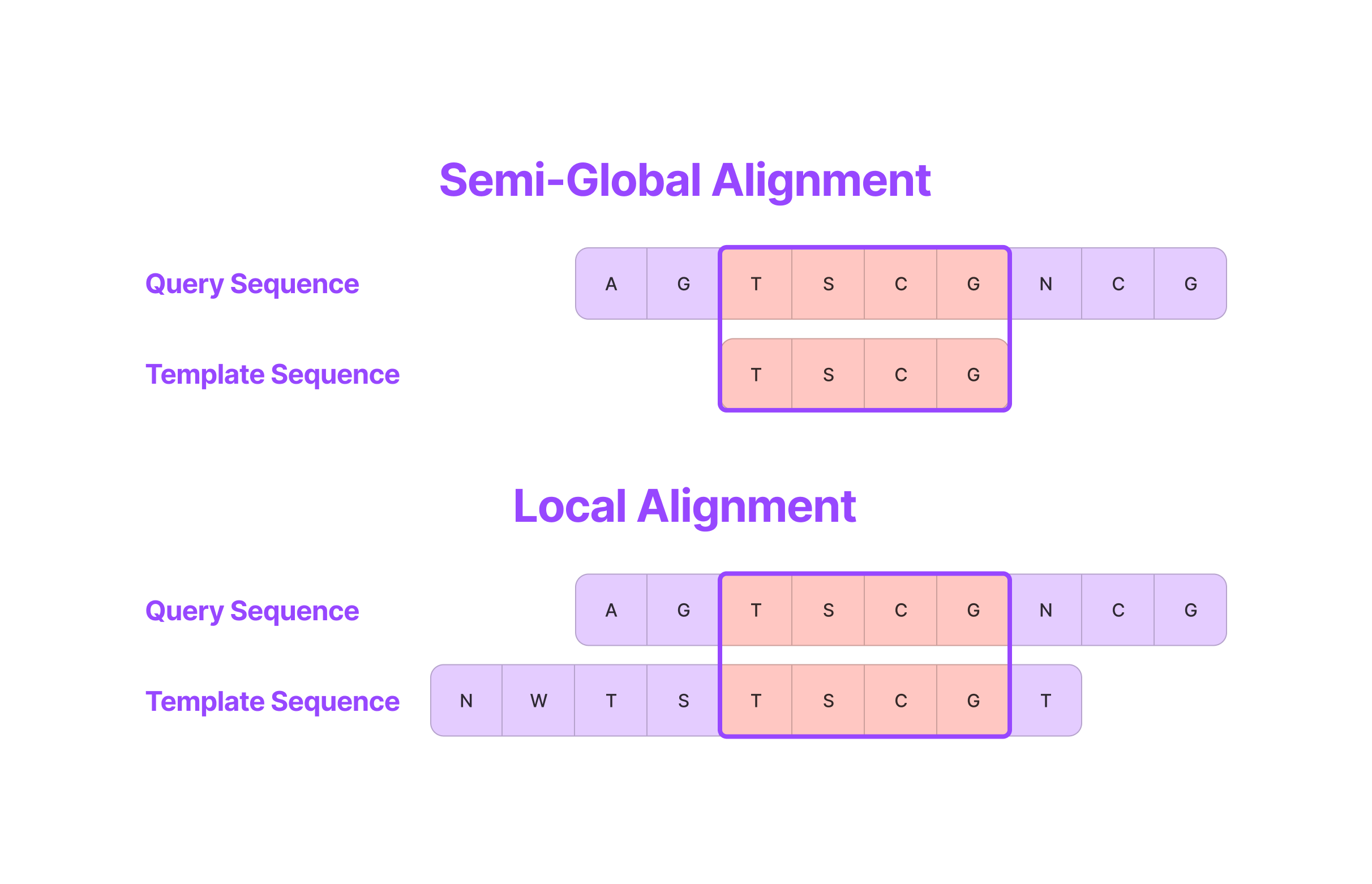







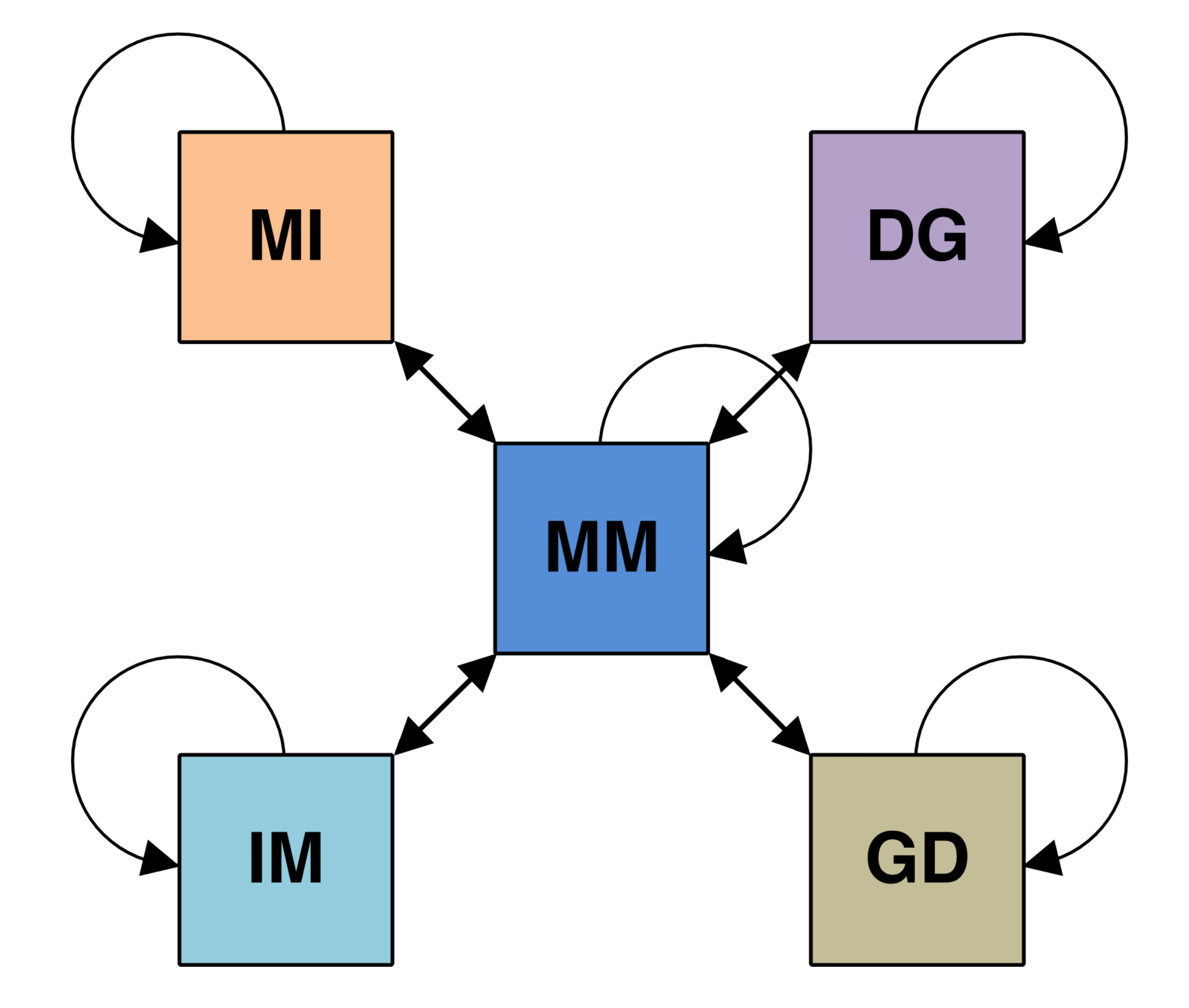


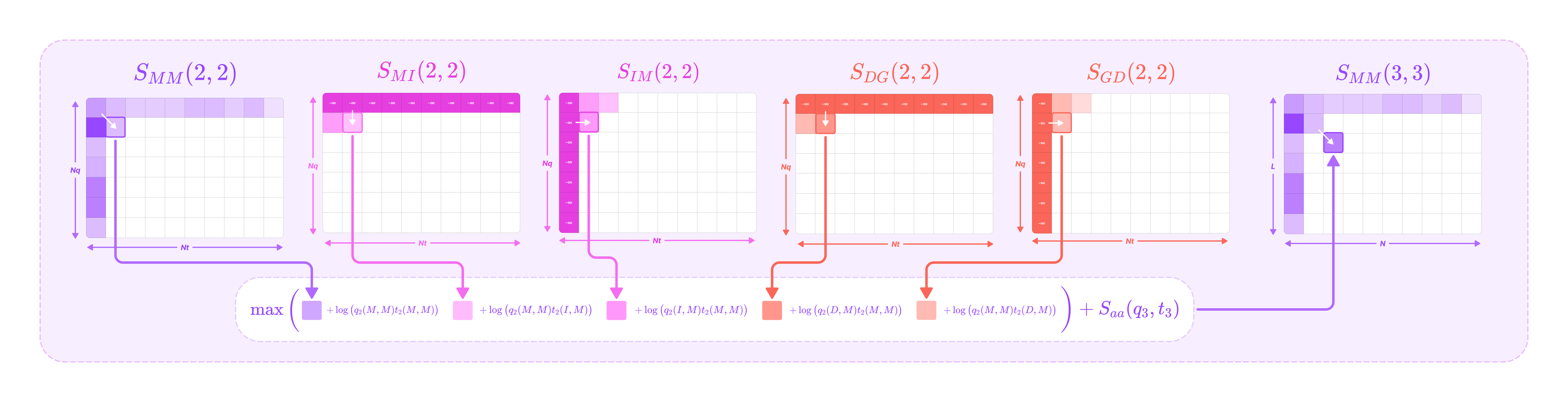




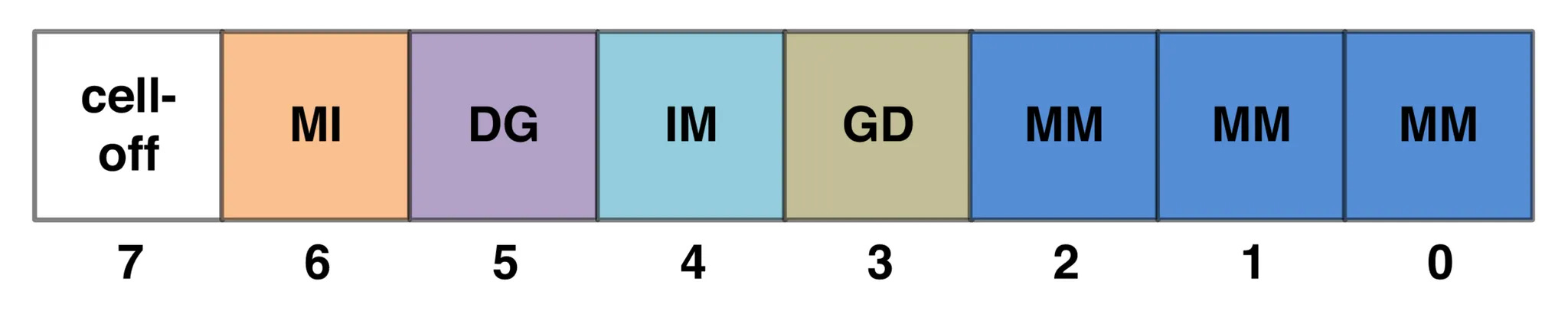
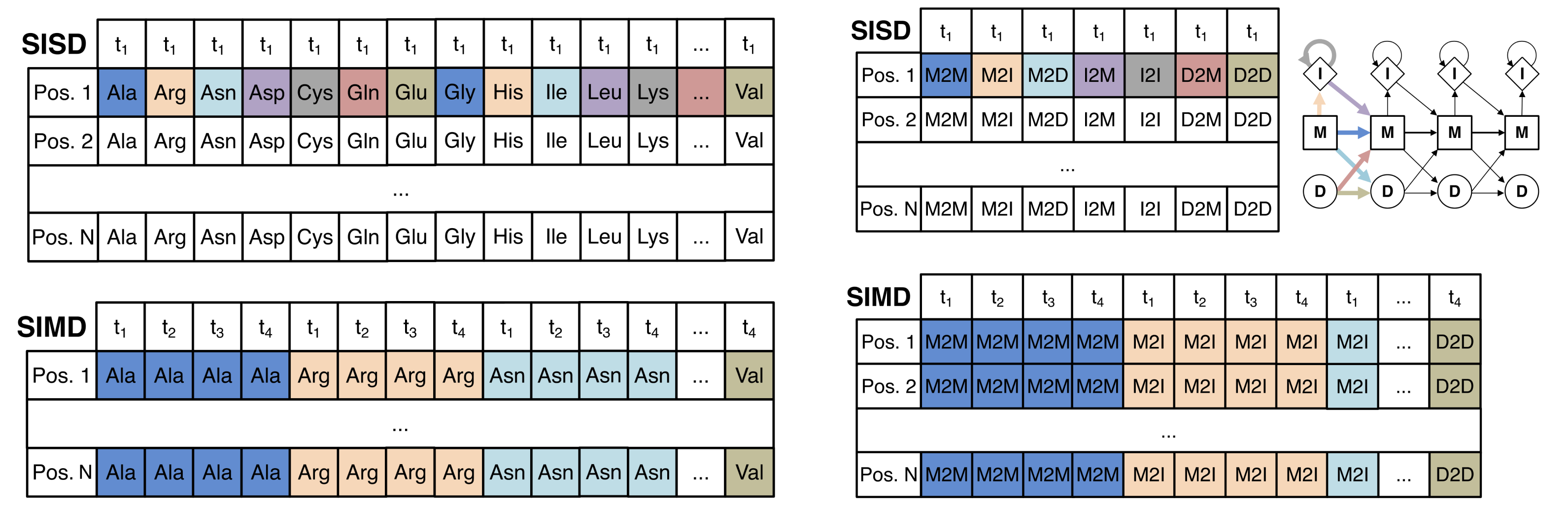





































































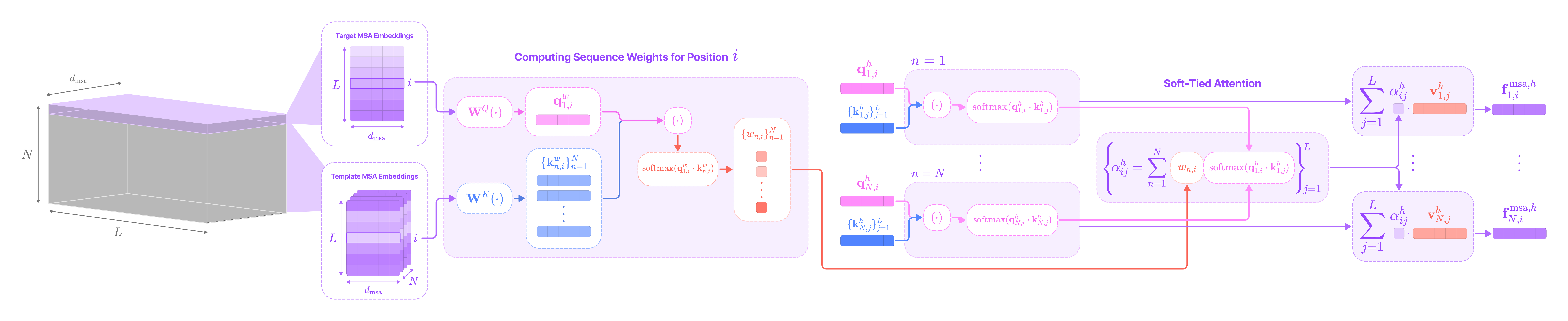








































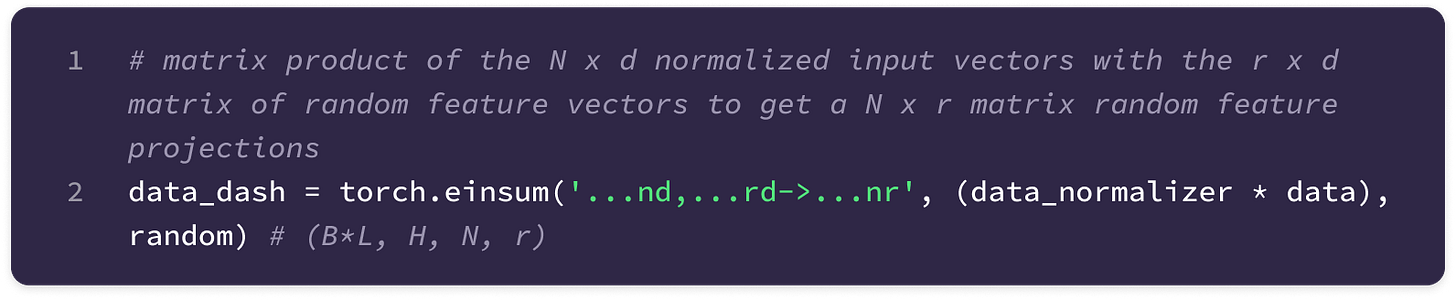












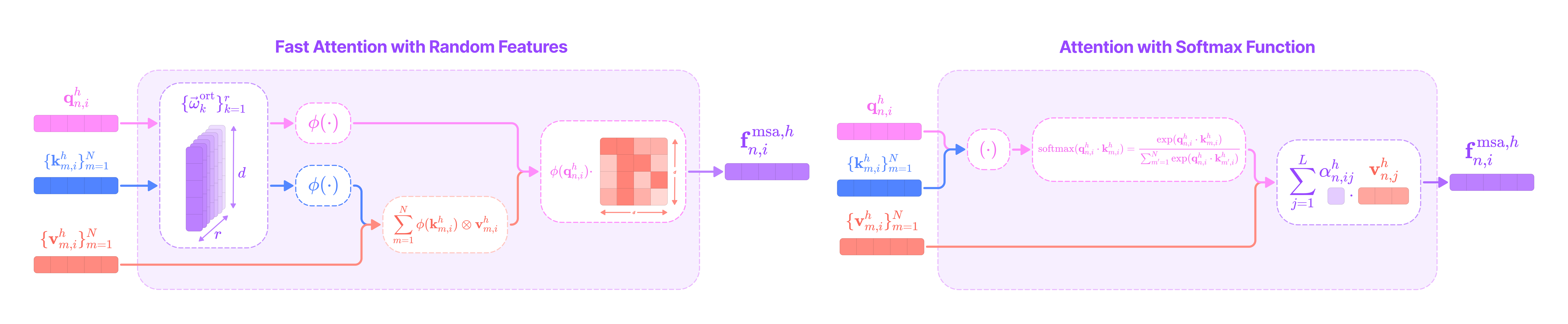




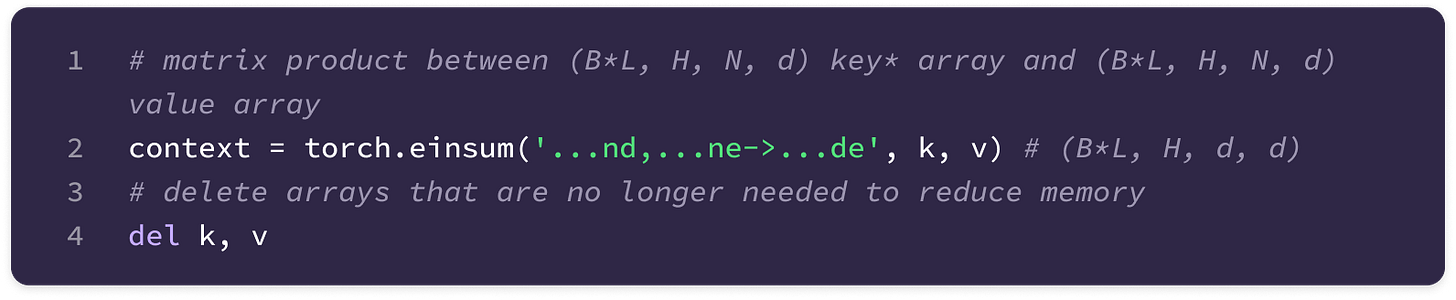


















Wow! I'm honestly scared to dive into it because just the substack article nearly froze my Google Chrome window. 🤣
How long did it take for you to write this?