



MolTrans: Transformers for Drug-Target Interaction Prediction
Comprehensive, visual breakdown of the MolTrans model and Transformer encoders for drug-target interaction (DTI) prediction.

90% of drugs fail during clinical trials.
Of these failures, 40-50% are caused by a lack of clinical efficacy, 30% are caused by toxicity, and 10-15% are caused by poor drug-like properties.

But only around 6 drug candidates make it to Phase I of clinical trials. The clinical phases are meant to confirm the candidates' efficacy, toxicity, and dosage, so why are so many drugs failing after passing pre-clinical testing?
Researchers screen hundreds of thousands of compounds using high-throughput screening (HTS) to find ‘hits’ that have a promising interaction with the target. HTS provides an optical measurement based on the strength of the interaction between an array of candidates against the protein target.
However, only 0.1 to 1 percent of candidates qualify as a ‘hit’, making HTS ineffective and expensive. Furthermore, this method doesn’t provide any information about the mechanisms of action and off-target effects of the candidates, resulting in many false positives and failures in future phases.
This is where drug-target interaction (DTI) prediction comes in.
DTI prediction enables scientists to identify promising compounds that interact with the target protein in silico, significantly reducing the time and cost associated with high-throughput screening. DTI prediction can be applied to:
Demystify the mechanisms of action of the drug — high-throughput screening provides limited insights into the specific protein domains that a drug candidate interacts with.
Predict interactions of drug candidates against similar proteins to evaluate off-target effects.
Screen existing drugs against other therapeutic targets for drug repurposing.
MolTrans: Overview of Architechture
MolTrans is a binary classification model that generates a probability of interaction between a protein and a small molecule drug. MolTrans was the first DTI model to incorporate Transformer encoder layers to derive complex interaction motifs between protein and drug substructures, achieving superior predictive performance on known and unseen drug-protein pairs.
At a high level, MolTrans (1) converts raw protein and drug inputs into sequences of highly recurring substructures (2) generates a contextual embedding that captures the chemical relationships between substructures in the given protein and drug molecule sequence and (3) generates a heatmap that represents the strength of interactions between the protein and drug substructures to predict the probability of interaction.

Sub-structure Decomposition
MolTrans uses a Frequent Conservative Subsequence (FCS) mining module to convert the sequence of amino acids representing a protein or SMILES characters representing a drug into sequences of fundamental biochemical substructures. Instead of learning interaction patterns between entire drugs and proteins, MolTrans learns from substructure interactions that are transferable to unseen drug and protein inputs.
The raw input of the drug is in the form of a Simplified Molecular Input Line Entry System (SMILES) string, which represents atoms and bonds in the format of a sequence of characters (e.g. C represents a carbon atom and = represents a double bond).
The raw input of the protein is in the form of sequences of characters representing the 20 amino acids.
The Frequent Conservative Subsequence (FCS) algorithm takes the following values as input:
V → the vocabulary of amino acid/SMILES tokens initialized with the 20 amino acids and SMILES characters.
W → tokenized set of all proteins/drugs in the databases where each token is initialized as individual atoms/amino acid symbols.
θ → frequency threshold that determines the number of occurrences that a pair of consecutive tokens must appear in W for it to be added to the vocabulary set V.
ℓ → the maximum size of the vocabulary set.
The FCS algorithm then updates V and W as follows:
The algorithm scans W for the most frequent pair of consecutive tokens (A, B).
Based on the frequency of (A, B):
frequency < θ → the algorithm stops and the updated vocabulary set V and tokenized set W are returned as output.
frequency ≥ θ → all instances of (A, B) in W are replaced with (AB) and (AB) is added to the vocabulary set V.
This algorithm continues to loop until V reaches the predefined maximum size ℓ or the frequency of the most frequent pair of tokens does not reach the threshold.
The substructure tokens generated by FCS are similar across datasets with differing characteristics, indicating that these substructures represent fundamental functional units of proteins and drugs. With θ set to 500, the FCS algorithm generated a total of 23,532 drug sub-structures from the ChEMBL dataset and 16,693 protein substructures from the UniProt dataset.
The final updated set W contains all the proteins and drugs in the dataset represented as sequences of these fundamental substructures.
After being fed into the FCS algorithm, the raw amino acid sequences and SMILES strings are converted into sequences of protein and drug substructures from the set V.
For the embedding and transformer modules, I will explain how the operations are performed on a protein input, but the same operations are applied to the drug input.
Embedding Module
Before entering the transformer module, the sequences of protein and drug substructures are converted into independent embedding matrices that captures the independent properties (content embedding) and positional information (positional embedding) of each protein and drug substructure.
Content Embedding
First, the sequence of protein substructures is converted into a one-hot encoding matrix with 16,693 rows (total number of protein substructures in V) and 545 columns (maximum length of protein sequence). The ith column of the matrix is a one-hot encoding vector with a value of 1 at the row corresponding to the ith protein substructure and zeros for all remaining entries.
Similarly, the sequence of drug substructures is converted into a 23,532 x 50 matrix where the jth column is a one-hot encoding vector with a value of 1 at the row corresponding to the jth drug substructure and zeros for all remaining entries.

Then, the one-hot encoding of the protein is multiplied by a learnable dictionary lookup matrix with dimensions 384 (length of embedding) x 16,693, where each column is a 384-dimensional content embedding vector that captures the independent properties of the substructure (e.g. binding sites, active sites, etc.) at the specified index corresponding to the row of the one-hot encoding. This gives a content embedding matrix for the protein composed of its constituent substructures as sequentially stacked content embeddings.
Positional Encoding
In a transformer model, every substructure of the protein flows through the model simultaneously so the model has no intrinsic knowledge of the relative positions of the substructures in the whole protein. Thus to allow the model to account for the relative positions when generating the final embedding, we must incorporate a way for the model to derive the position of a given substructure embedding relative to other substructures.
The positional encoding is a vector with the same dimension as the content embedding (d = 384) that is unique for every position in the protein sequence. For a substructure at position t in the protein, the positional encoding (p_t) is defined as follows:
where ωₖ is defined as:
At a high level, the positional encoding is a vector of pairs of sine and cosine functions of decreasing frequency along the vector dimension.

The primary advantage of such an embedding is that it allows the model to easily identify the relative distance of a given substructure with the substructure that it is being attended to in the self-attention module. This is because the positional encoding p_(t+n) for a substructure at a fixed distance n from the given substructure at position t can be obtained by applying a linear transformation, M (where M does not depend on t) on the positional encoding p_t.
where M is defined as:
For a deeper understanding of positional encodings and the derivation of the matrix M above, check out Amirhossein Kazemnejad's positional embedding tutorial.
Finally, the positional encoding is added to the content embedding to obtain the final embedding to be fed as input into the transformer module.
Transformer Module
MolTrans incorporates two transformer encoder blocks for proteins and drugs, each with a self-attention layer consisting of 12 attention heads and a feed-forward network with intermediate dimension of 1536.
Recently, there has been a rise in deep language models for protein prediction tasks given their ability to learn rich feature representations from large unstructured sequence datasets.
The Transformer model, introduced in 2017 by Vaswani et al., has been widely adopted for natural language processing (NLP) and protein prediction tasks given its ability to learn relationships between distant tokens (words or amino acids) across the entire sequence (sentences or proteins) using the attention mechanism.
In the context of protein prediction tasks, attention can generate a vector representation of a given substructure in a protein that is a weighted sum of the vector representations of all the other substructures in the protein. The weights model the chemical relationships between substructures in the protein.
Transformer models for protein prediction tasks consist of two overarching steps:
The encoder takes large sets of protein sequences of various lengths as input, learns the relevant features from the protein sequences that are required to solve the specified prediction problem, and produces representations of all the protein sequences as fixed-length real (floating-point) vectors containing these necessary features called internal learned representations.
Then, the internal learned representation is fed into a conventional neural network (e.g. convolutional layers, fully connected layers) to generate a classification or regression prediction.
MolTrans leverages transformer encoder layers to derive an internal learned representation of the protein and drug molecule that takes into account the complex chemical relationships between the protein and molecule substructures.
An encoder block consists of four components: self-attention, feed-forward network, residual connection, and layer normalization.

Self-Attention for a Single Sub-structure
Consider a single embedding vector representing a single sub-structure in a protein (we will call this the “present substructure”). We will walk through how this embedding vector is converted into the final embedding.
First, the embedding vector is converted into three vectors: a query (q) vector, a key (k) vector, and a value (v) vector.
These vectors have dimension 32 whereas the embedding vector has dimension 384. The dimension (which we will call the attention head size) is set to be the embedding dimension divided by the number of attention heads (384 ÷ 12 = 32) to ensure that multi-head attention (more on this later) does not increase computational cost.
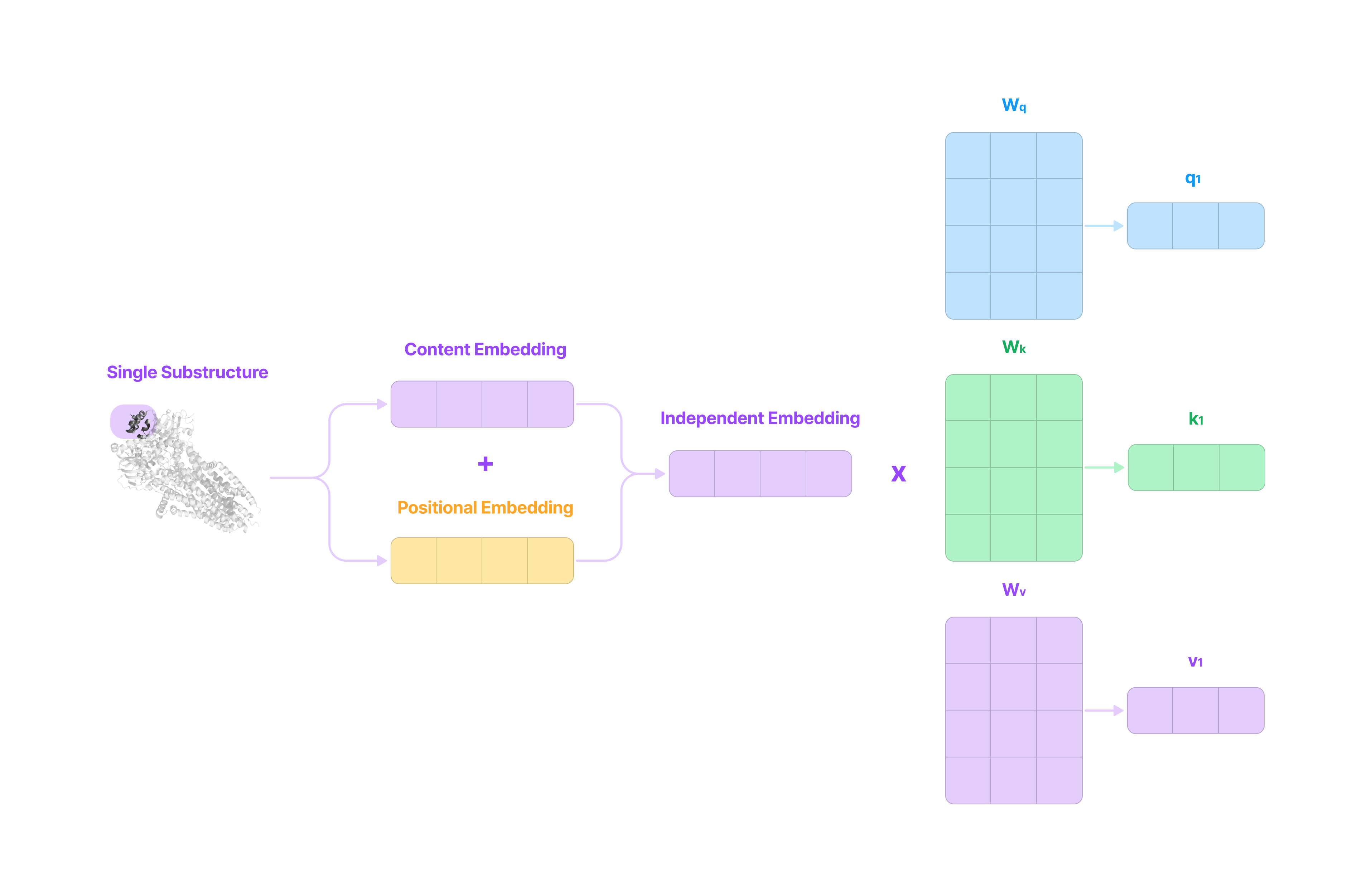
These vectors are generated by multiplying the embedding vector with the corresponding 384 x 32 query (Wq), key (Wk), and value (Wv) weight matrices optimized during the training process.
After the three vectors are generated for the present substructure and every other substructure in the protein, the attention layer will generate a score (value between 0 and 1) for every other substructure representing the strength of the chemical relationship between that substructure and the present substructure.
Each score is generated in the following steps:
The scalar dot product of the present substructure's query vector and the other substructure's key vector is calculated.
The dot product is divided by the square root of 32 (attention head size). This prevents the dot product from growing too large and pushing the gradient to zero when the softmax function is applied. Gradients of zero can result in a vanishing gradient when calculating gradients in previous layers of the model using the chain rule during gradient descent, preventing the model from learning effectively.

For large values, the gradient (or slope) of the softmax function approaches zero. (Source) A softmax function is applied to convert the output of Step 2 into a value between 0 and 1 such that all the scores for the given substructure sum up to 1 (learn more about why the softmax function works here).

Diagram showing how the softmax function transforms a vector (which is a set of dort products corresponding to every protein substructure) into a vector of scores between 0 and 1 such that all the scores sum to 1. (Source)


Intuitively, the score is higher when the pair of substructures have a strong chemical relationship and near zero when the chemical relationship between the substructures is irrelevant in the current representation subspace (more on this later).
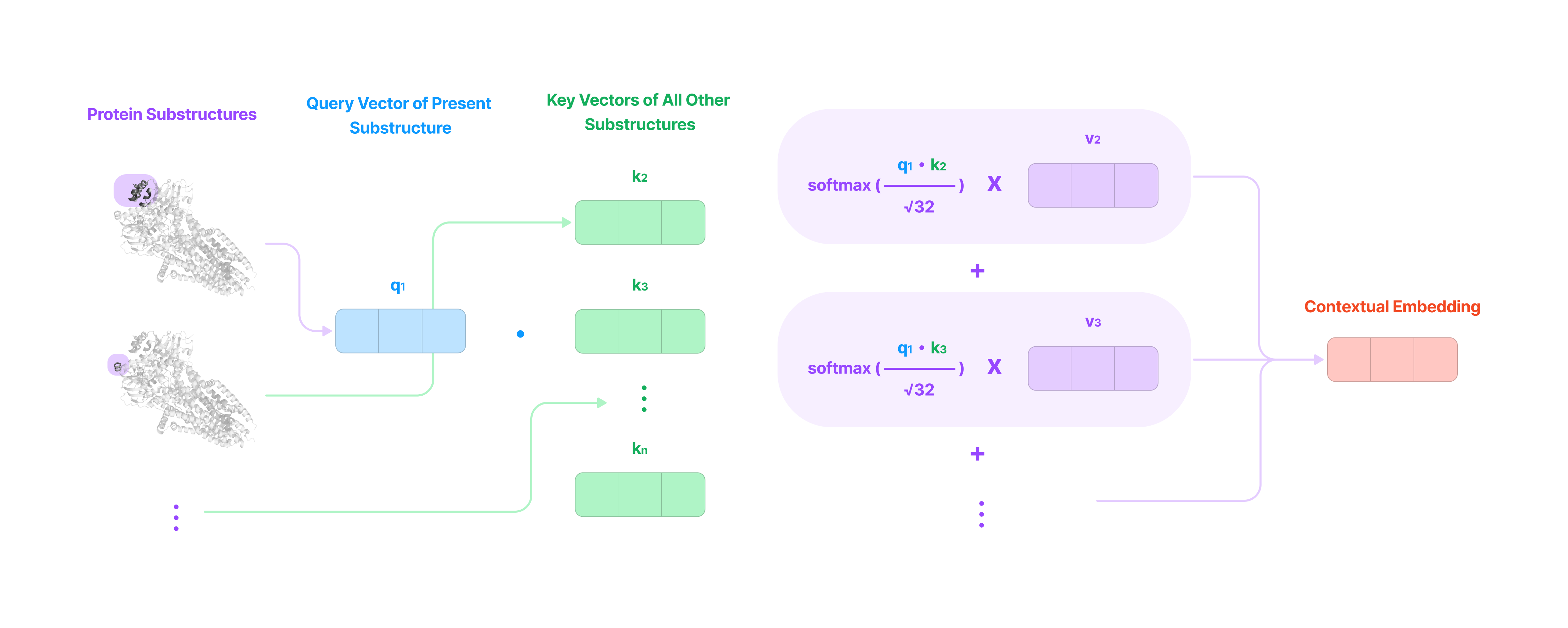
Every value vector representing every substructure in the protein is multiplied by its corresponding score and the sum of all the weighted value vectors becomes the contextual embedding of the present substructure.
Self-Attention on a Protein using Matrices
Now that we understand how self-attention works on a single substructure in a protein, we can generate the contextual embedding for the full protein by following the same steps on a 384 x 545 (maximum length of protein sequence) matrix where each row is the independent embedding of a single substructure.

The Wq/Wk/Wv matrices transform the input embedding into 32 x 545 query (Q), key (K), and value (V) matrices.
The query matrix is multiplied by the transpose of the key matrix.
Every element of the matrix is divided by √32 and the softmax function is independently applied to every row.
The value matrix is multiplied by the 545 x 545 matrix of scores to produce the contextual embedding of the full protein.
Similarly, the independent substructure embeddings of the small molecule drug are packed into a 384 x 50 (maximum length of drug sequence) matrix, and the same operations are performed.
Multi-Head Attention
We have described the function of a single attention ‘head’ which can capture a single ‘representation subspace’ of the protein. However, a protein or drug can have diverse chemical relationships that require multiple representation subspaces.
Thus, Transformer models leverage multi-head attention, where attention mechanisms are performed in parallel which project the input protein embedding into distinct representation subspaces that each capture a particular type of relationship between protein substructures.
MolTrans uses 12 attention heads, each of which projects the input embedding to a smaller 32-dimensional feature space (attention head size) so that the total computational cost of multi-head attention is similar to that of fully-dimensional single-head attention.
During training, each attention head optimizes a set of randomly initialized Wq/Wk/Wv weight matrices. This produces 12 unique sets of weight matrices that project the input embedding to 12 distinct sets of Q/K/V matrices and produce 12 contextual embedding matrices in different representation subspaces.

These embedding matrices are concatenated horizontally and multiplied by an additional 384 x 384 weight matrix, which is trained to learn the relative importance of each representation subspace in describing the protein. The final output is a single 545 x 384 contextual embedding matrix that captures all the diverse chemical relationships between substructures.

Here’s the implementation of the multi-head self-attention mechanism in PyTorch (the full code for the MolTrans model can be found here):
class SelfAttention(nn.Module):
"""Annotated version of the code taken from https://github.com/kexinhuang12345/MolTrans/tree/master"""
def __init__(self, hidden_size, num_attention_heads, attention_probs_dropout_prob):
super(SelfAttention, self).__init__()
# makes sure that the hidden_size (dimension of embedding) is a multiple of the number of attention heads
assert hidden_size % num_attention_heads == 0
self.num_attention_heads = num_attention_heads
# size of each attention head is 32
self.attention_head_size = int(hidden_size / num_attention_heads)
# total size of all attention heads is 384 which is the same as hidden_size
self.all_head_size = self.num_attention_heads * self.attention_head_size
# define linear transformations that project input embedding to query, key and value tensors
self.query = nn.Linear(hidden_size, self.all_head_size)
self.key = nn.Linear(hidden_size, self.all_head_size)
self.value = nn.Linear(hidden_size, self.all_head_size)
self.dropout = nn.Dropout(attention_probs_dropout_prob)
# function that splits the query, key, and value tensors into 12 attention heads
def transpose_for_scores(self, x):
new_x_shape = x.size()[:-1] + (self.num_attention_heads, self.attention_head_size)
x = x.view(*new_x_shape)
return x.permute(0, 2, 1, 3)
def forward(self, hidden_states, attention_mask):
# project input embedding into combined query, key, and value tensors with shape (batch_size, seq_length, all_head_size)
mixed_query_layer = self.query(hidden_states)
mixed_key_layer = self.key(hidden_states)
mixed_value_layer = self.value(hidden_states)
# split the tensors to multiple heads with shape (batch_size, num_attention_heads, seq_length, attention_head_size)
query_layer = self.transpose_for_scores(mixed_query_layer)
key_layer = self.transpose_for_scores(mixed_key_layer)
value_layer = self.transpose_for_scores(mixed_value_layer)
# take the dot product between query and key to get the raw attention scores
attention_scores = torch.matmul(query_layer, key_layer.transpose(-1, -2))
# divide by square root of 32
attention_scores = attention_scores / math.sqrt(self.attention_head_size)
# mask applied on padding tokens
attention_scores = attention_scores + attention_mask
# convert the attention scores to probabilities using softmax
attention_probs = nn.Softmax(dim=-1)(attention_scores)
# applies dropout to the attention probabilities tensor by randomly setting probs to zero, which prevents the model to become overly reliant on specific positions in the sequence
attention_probs = self.dropout(attention_probs)
# scale value tensor by attention probabilities
context_layer = torch.matmul(attention_probs, value_layer)
# context vectors from all attention heads are concatenated to form a single output tensor with same dimensions as the input embedding matrix
context_layer = context_layer.permute(0, 2, 1, 3).contiguous()
new_context_layer_shape = context_layer.size()[:-2] + (self.all_head_size,)
context_layer = context_layer.view(*new_context_layer_shape)
return context_layerResidual Connection and Layer Normalization
Following each multi-head attention layer module and fully connected layer, there is a residual connection and layer normalization step.

A residual connection (or skip connection) is performed by adding the input embedding matrix to the output embedding matrix. Residual connections allow the model to retain information from the original embedding and prevent vanishing gradients by introducing an addition operation for the gradient to skip past layer operations during backpropagation.
Layer normalization transforms the features of the embedding to have a mean of 0 and variance of 1, to stabilize the distribution of features across layers for smoother gradient flow.
The following SelfOutput class is applied after the multi-head attention:
# layer normalization class
class LayerNorm(nn.Module):
def __init__(self, hidden_size, variance_epsilon=1e-12):
super(LayerNorm, self).__init__()
self.gamma = nn.Parameter(torch.ones(hidden_size))
self.beta = nn.Parameter(torch.zeros(hidden_size))
self.variance_epsilon = variance_epsilon
def forward(self, x):
# calculate mean of x
u = x.mean(-1, keepdim=True)
# calculate variance of x
s = (x - u).pow(2).mean(-1, keepdim=True)
# normalize x by subtracting the mean and dividing by the square root of the variance plus the small constant
x = (x - u) / torch.sqrt(s + self.variance_epsilon)
# scale and shift by learnable parameters
return self.gamma * x + self.beta
# applied to the output of the multi-head self-attention layer
class SelfOutput(nn.Module):
def __init__(self, hidden_size, hidden_dropout_prob):
super(SelfOutput, self).__init__()
self.dense = nn.Linear(hidden_size, hidden_size)
self.LayerNorm = LayerNorm(hidden_size)
self.dropout = nn.Dropout(hidden_dropout_prob)
def forward(self, hidden_states, input_tensor):
# fully-connected layer
hidden_states = self.dense(hidden_states)
# applies dropout before residual connection
hidden_states = self.dropout(hidden_states)
# residual and layer normalization step
hidden_states = self.LayerNorm(hidden_states + input_tensor)
return hidden_states Feed-Foward Network
While the self-attention mechanism applies dependencies between the different substructures in the protein or drug, the feed-forward network (FFN) that follows the self-attention layer is applied independently on each substructure embedding.
The FFN transforms each contextual substructure embedding vector (x) with the following steps:

The first fully connected layer transforms the 384-dimensional vector representing a single substructure to a higher-dimensional vector (dimension 1536 used in MolTrans) by multiplying the vector by a 384 x 1536 weight matrix and adding a 1536-dimensional bias vector.
\(x_2 = x_1W_{1}+b_1 \)A non-linear ReLU activation function is applied element-wise to the higher-dimensional embedding vector. For each element of the embedding vector, the ReLU function outputs 0 for negative values or itself for positive values.
\(x_3=max(0, z_2)\)The second fully connected layer projects the 1536-dimensional vector back to its original dimension (384) by multiplying it with a 1536 x 384 weight matrix and adding a 384-dimensional bias vector.
\(x_4=x_3W_2+b_2\)
The full mechanism of the FFN can be written as:
The purpose of the FFN in an encoder block is three-fold:
Since self-attention layers are primarily linear (consisting of matrix multiplication and computing weighted sums of vectors), the FFN introduces non-linearity to the encoder that allows the model to learn more complex relationships in the data that self-attention cannot capture.
Projecting the embedding to a higher dimension allows the model to learn features in a richer representation space.
The FFN is applied after the residual connection that reminds the model of the original embedding and is applied independently to each substructure, allowing the model to enhance the independent characteristics of each substructure that may be lost in the self-attention layers.
Here are the classes that implement the feed-forward network and the following residual and normalization steps in PyTorch:
# linear transformation from hidden_size of 384 to intermediate_size of 1536
class Intermediate(nn.Module):
def __init__(self, hidden_size, intermediate_size):
super(Intermediate, self).__init__()
self.dense = nn.Linear(hidden_size, intermediate_size)
def forward(self, hidden_states):
hidden_states = self.dense(hidden_states)
hidden_states = F.relu(hidden_states)
return hidden_states
# linear transformation back to hidden_size with additional residual + layer normalization step
class Output(nn.Module):
def __init__(self, intermediate_size, hidden_size, hidden_dropout_prob):
super(Output, self).__init__()
self.dense = nn.Linear(intermediate_size, hidden_size)
self.LayerNorm = LayerNorm(hidden_size)
self.dropout = nn.Dropout(hidden_dropout_prob)
def forward(self, hidden_states, input_tensor):
hidden_states = self.dense(hidden_states)
hidden_states = self.dropout(hidden_states)
hidden_states = self.LayerNorm(hidden_states + input_tensor)
return hidden_statesFull Transformer Module
Now that we have covered the mechanism of each component of the transformer module, here is a reminder of what it looks like when all the layers come together:

So far, the MolTrans model has generated two matrix embeddings representing the protein and the drug molecule independently. It’s time to dive into how the model combines these two matrices to generate a prediction on whether an interaction occurs.
Interaction Module
The interaction module consists of two components: a pairwise interaction operation that generates a matrix representing the strength of every drug substructure with every protein substructure and a neighborhood interaction layer that uses a convolutional neural network (CNN) layer to extract local interaction motifs.
Pairwise Interaction
The product of the protein embedding matrix and the transpose of the drug embedding matrix gives a pairwise interaction matrix (𝐈), where the rows represent the strength of interaction of a single drug substructure with every protein substructure and the columns represent the strength of interaction of a single protein substructure with every drug substructure.
𝐈 is a 50 (maximum length of a drug sequence) by 545 (maximum length of a protein sequence) matrix where the value at position (i, j) is the dot product of the ith substructure of the drug and the jth substructure of the protein.

Since the dot product is a measure of the proximity between two vectors, a larger dot product corresponds to a stronger interaction between the two substructures.

Neighborhood Interaction
Rather than a single protein substructure interacting with a single drug substructure, drug-protein interactions are often triggered by a combination of interactions of nearby protein and drug substructures. Thus, a convolutional neural network (CNN) layer is applied to the pairwise interaction matrix 𝐈 to capture patterns in the collective interaction between neighborhood substructures. This allows the model to learn patterns in neighboring substructure interactions that trigger stronger or weaker overall interactions between the full protein and drug.
A CNN leverages small 2-dimensional matrices, called filters or kernels, containing weights that can detect neighborhood interaction motifs, such as hydrogen bonds, hydrophobic interactions, and other local interaction patterns. The same filter (matrix of weights) slides over the entire input matrix and calculates a value at each position, detecting a specific motif regardless of where it occurs within the drug-protein interaction space.
The weights of a filter are trained to learn a motif that recurs frequently in positive drug-target interactions, and multiple filters can be trained to detect several distinct motifs.

The MolTrans model applies three 3 x 3 filters, each of which transforms the pairwise interaction matrix into a separate feature map with the following operations starting from the top-left corner of 𝐈:
The 3 x 3 subset of the input matrix is multiplied element-wise with the filter weights.
All nine products are summed to produce the top-left value of the feature map.
The filter is then shifted to the right by one column and Steps 1 and 2 are repeated.
After the filter reaches the top-right corner, it shifts down by one row and the process repeats from right to left.
This repeats until the filter reaches the bottom-right corner.

Since MolTrans does not apply padding, the CNN layer produces three feature maps that each have dimensions 543 x 48.
Here’s the implementation of the CNN layer in PyTorch:
self.icnn = nn.Conv2d(1, 3, 3, padding = 0)Finally, all three feature maps are flattened into a 78192-dimensional vector and fed through a decoder module consisting of a series of fully connected layers (with trainable weight matrices and bias vectors), ReLU activation functions, and batch normalization steps that transform the mean to 0 and variance to 1 for each feature across a batch of training examples.
Here’s the implementation of the decoder in PyTorch:
self.decoder = nn.Sequential(
nn.Linear(self.flatten_dim, 512), #converts dim from 78192 to 512
nn.ReLU(True), #applies ReLU activation element-wise
nn.BatchNorm1d(512), #normalization
nn.Linear(512, 64), #converts dim from 512 to 64
nn.ReLU(True), #applies ReLU activation element-wise
nn.BatchNorm1d(64), #normalization
nn.Linear(64, 32), #converts dim from 64 to 32
nn.ReLU(True), #applies ReLU activation element-wise
#output layer
nn.Linear(32, 1) #outputs the probability of interaction
)The final output of the decoder is the probability of an interaction (value between 0 and 1) between the protein and small molecule drug.
Finally, we have reached the end of the model. Now, let’s dive into how MolTrans is trained.
Training
All the weights and biases from the embedding module, Transformer module, and interaction module are jointly trained using binary cross-entropy loss and optimized using the Adam optimizer. The MolTrans model is trained on BIOSNAP, DAVIS, and BindingDB datasets.
Binary Cross-Entropy Loss Function
To measure the model's predictive performance and calculate the gradients for gradient descent, a binary cross-entropy (BCE) loss function is used:
where p is the predicted probability of interaction generated by the model and y is the true label (either 0 for no interaction or 1 for an interaction).
For training examples with a positive label (y = 1), the loss is equal to -log(p). Intuitively, this works because we want the loss to be small for values of p close to 1 and large for predictions close to zero.
Conversely, for training examples with a negative label (y = 0), the loss is equal to -log(1-p), which gives a large loss for values of p close to 1 and a small loss for values close to zero.

BCE loss is a great choice for binary classification tasks because it is continuous and differentiable, producing gradients that are efficient in minimizing the function.
Gradient Descent with Adam Optimizer
MolTrans uses the Adam optimizer with learning rate 1e-5 and a batch size of 64. The Adam optimizer is a stochastic gradient descent algorithm that adapts the step size for each iteration (epoch) and parameter, enabling stable updates and faster convergence.
Gradient descent is an algorithm that iteratively updates the weights and biases (parameters) of the model to minimize the error calculated by the loss function. At a high level, a gradient descent algorithm:
Calculates the partial derivative (which we will call the gradient from now on) with respect to (w.r.t.) every parameter using the chain rule. The partial derivatives of every parameter in the model make up the gradient of the loss function.
For a deeper understanding of the calculus behind backpropagation, see 3Blue1Brown’s video on how gradients are calculated in a simple neural network.
Updates each parameter by subtracting a step size, which is usually some variation of the gradient * learning rate. This adjusts the parameter in the direction that minimizes the loss function. In stochastic gradient descent, the partial derivatives of the loss function w.r.t. a parameter are calculated for a specified batch size of training examples (instead of the entire training set like in standard gradient descent) and summed before being multiplied by the learning rate (value between 0 and 1).
Repeats until the loss function converges to zero or a value close to zero.
The Adaptive Moment Estimation (Adam) optimizer is a stochastic gradient descent algorithm that adapts the step size at each iteration (epoch) based on the first moment (moving average) and second moment (variance) of the gradients of each parameter.
Instead of the step size being the gradient * learning rate, Adam updates each parameter by a step size that is adjusted by the first and second moment of the parameter:
Let’s break down the elements of the step size for the Adam optimizer:
The first moment or moving average at the current iteration (m_t) of each parameter is calculated by taking an exponentially decaying average of all the past gradients w.r.t. that parameter. The equation for the first moment for a single parameter is:
\(m_t=β_1\cdot m_{t−1}+(1−β_1)\cdot g_t \)where β_1 is the exponential decay rate, m_(t-1) is the average of past gradients, and g_t is the gradient of loss function w.r.t. the parameter at the current iteration.
β_1 (typically set to 0.9 by default) determines how much weight is given to past gradients compared to the gradient at the current iteration. Since β_1 is set close to 1, the average is heavily weighted towards the values of past gradients with a small weight given to the current gradient.
Instead of multiplying the learning rate directly with the gradient, it is multiplied by the first moment to smooth out short-term fluctuations or noise in the gradients to ensure stable updates to the parameter that reflect the overall trend of the gradient.
The learning rate (η) is the default learning rate that will be adjusted based on the second moment.
The second moment or variance at the current iteration (v_t) of each parameter is calculated by taking an exponentially decaying average of the squares of all the past gradients w.r.t. that parameter. The equation for the second moment of a single parameter is:
\(v_t=β_2\cdot v_{t−1}+(1−β_2)\cdot g_t^2\)where β_2 is the exponential decay rate, v_(t-1) is the average of past variances, and gₜ is the gradient of loss function w.r.t. the parameter at the current iteration.
Similarly to the first moment, β_2 (typically set to 0.999 by default) determines how much weight is given to past variances compared to the variance at the current iteration. Since β_2 is set close to 1, the variance is heavily weighted towards past variances, making the model less sensitive to sudden changes in gradient magnitude.
The second moment indicates how much the gradients of a parameter fluctuate over past iterations. The learning rate is divided by the square root of the second moment so that parameters with high variance (greatly fluctuating gradients) have smaller learning rates to prevent overshooting and parameters with low variance (stable gradients) have higher learning rates to accelerate convergence.
Since the first and second moments are initialized to zero and the decay rates are close to 1, the values for m_1 and v_1 are biased towards zero. Thus, m_t and v_t are divided by a bias correction term that starts at a small value and approaches 1 with each iteration, reducing the significance of the correction.
\(\hat{m}_t=\frac{m_t}{1−β_1^t}\text{, } \hat{v}_t=\frac{v_t}{1−β_2^t}\)ϵ is a small constant (default value of 1e-8) to prevent division by zero.
The Adam optimizer speeds up convergence, is robust to noisy gradients, and eliminates the need for manual hyperparameter tuning.

The following code defines an Adam optimizer that improves the .parameters() of the model, with a learning rate of 1e-5:
learningRate = 1e-5
optimizer = torch.optim.Adam(model.parameters(), lr=learningRate)Using the Adam optimizer with the learning rate set to 1e-5 and batch size of 64, MolTrans converges between 8 and 15 epochs.
Negative Edge Subsampling
Subsampling datasets for training is required due to unbalanced data. Instead of random subsampling used to train MolTrans, de la Fuente et al. proposed a subsampling strategy that improves predictive performance.
Since drugs don’t interact with most target proteins, DTI prediction tasks struggle from unbalanced training data, where the number of negative training examples (all non-interacting drug-protein pairs) is exponentially greater than the number of positive training examples (interacting drug-protein pairs).
MolTrans uses random subsampling to balance sparse positive cases with abundant negative cases. However, this method limits the number of difficult-to-classify negative cases the model is trained on and prevents the model from learning more subtle features that prevent a drug from interacting with a given protein.
Since interaction properties are preserved in a protein’s function rather than the specific amino acid sequence, de la Fuente et al. proposed a novel subsampling strategy to select high-quality negative examples based on the structural similarity of every other protein in the dataset to the target protein.
The structural similarity between two proteins is calculated as the Root Mean Square Deviation (RMSD) between the locations of the alpha carbon atoms (carbon connected to the functional group of every amino acid) in the protein backbone. The lower the RMSD between two proteins (calculated in Angstrom’s), the more structurally similar they are.

The strategy selects negative examples in the following steps for every positive drug-target interaction:
Find all negative drug-protein pairs with the same drug.
Calculate the RMSD between the target protein and all other proteins in the dataset with negative interactions with the drug.
Determine what to do with the drug-protein pair based on the RMSD value:
0-2 Å → discard example.
Proteins in this interval have an extremely low RMSD, likely because they are too small or simple and can align non-specifically with the target protein. Thus, they do not make good training examples.
2.5-5 Å → include in the validation set.
Proteins in this interval are very similar to the target protein but are labeled as 0 (non-targets). If included in the training data, they can introduce ambiguity and hinder the model’s ability to discern distinguishing features of target proteins. However, they can be used as challenging validation examples.
5-(6-20) Å → include in training set.
Proteins in this interval are similar enough to the target proteins to make the training challenging (helping the model learn nuanced differences) but different enough that they can confidently be labeled as non-targets (true negatives).

After retraining MolTrans with this strategy, de la Fuente et al. achieved superior AUC compared to the original random subsampling strategy when validated on BIOSNAP and BindingDB datasets.
Final Thoughts
MolTrans represents a significant advancement in inductive DTI prediction models, characterized by their ability to learn underlying protein and drug substructure interaction motifs and generalize to unseen protein and drug molecules. Inductive models generate valuable insights into which substructures are active during interactions. These insights can enable the rational design of drugs — from integrating substructures involved in interactions with a protein target to removing substructures from drug candidates involved in unwanted off-target effects — as well as the repurposing of existing drugs for new therapeutic targets that share active substructures found in the drug’s known target.
By leveraging the power of inductive deep learning models or DTI prediction, we can drastically shorten the drug discovery pipeline and bring life-saving treatments to patients faster than ever before.
Thank you for reading! This post was written for those looking to fully grasp the architecture and training design of a deep-learning model for DTI prediction. If you have any suggestions, please feel free to reach out via LinkedIn.
The diagrams were inspired by “The Illustrated Transformer” by Jay Alammar and “An Opinionated AlphaFold3 Field Guide” by Dimension Research.
Alchemy Bio is a blog where I share transformational ideas in computational biology, synbio, and biotech. If you don’t want to miss upcoming blogs, you should consider subscribing for free to have them delivered to your inbox: