

Designing the Next Generation of mRNA Therapeutics
A 20-minute walkthrough of our recent preprint, “mRNAutilus: Multi-Objective-Guided Discrete Generation of mRNA with Optimized Therapeutic Properties”


In 2020, two mRNA vaccines went from an untested sequence to a patient in under a year. For most of the history of medicine, a new vaccine took a decade. The mRNA platform didn’t just collapse that timeline; it introduced an entirely new class of therapeutic. mRNA therapeutics didn’t just deliver a SARS-CoV-2 spike protein, but proved that we can now manufacture a protein within a patient’s body on demand by giving their cells a transient blueprint.
Five years later, innovation in mRNA therapeutic platforms is expanding rapidly. There are now four FDA-approved mRNA therapeutics on the market, several more in Phase 3 trials for cancer and rare disease, and a growing pipeline of mRNA-encoded payloads that go far beyond classical proteins, ranging from prime editors, base editors, and antibodies to replacement enzymes and engineered intracellular degraders.
However, while the delivery system, the lipid nanoparticle (LNP), has gotten most of the attention1, the mRNA sequence itself has largely been engineered with tools that were designed in the 1980s.
This is the gap that we aim to close with mRNAutilus.2

The central question that we address is: how can we design every part of a therapeutic mRNA transcript, end-to-end, so the resulting sequence exhibits all the necessary properties of a successful therapeutic?
It is clear that generative modeling is the right tool to narrow down the search space, so let’s dive deep into how we tackle this with mRNAutilus, our multi-objective-guided masked discrete diffusion model for full-length mRNA design.
In this article, I break down what mRNA does in a therapeutic setting, how mRNAs are designed today, where current approaches fall short, and how mRNAutilus closes those gaps.
mRNA: A Versatile Therapeutic Modality
The cell’s blueprint for every protein
Messenger RNA (mRNA) is the middle step in the central dogma. DNA gets transcribed into mRNA by RNA polymerase, then mRNA gets translated into protein by the ribosome. Every protein your body has ever made passed through an mRNA intermediate first.
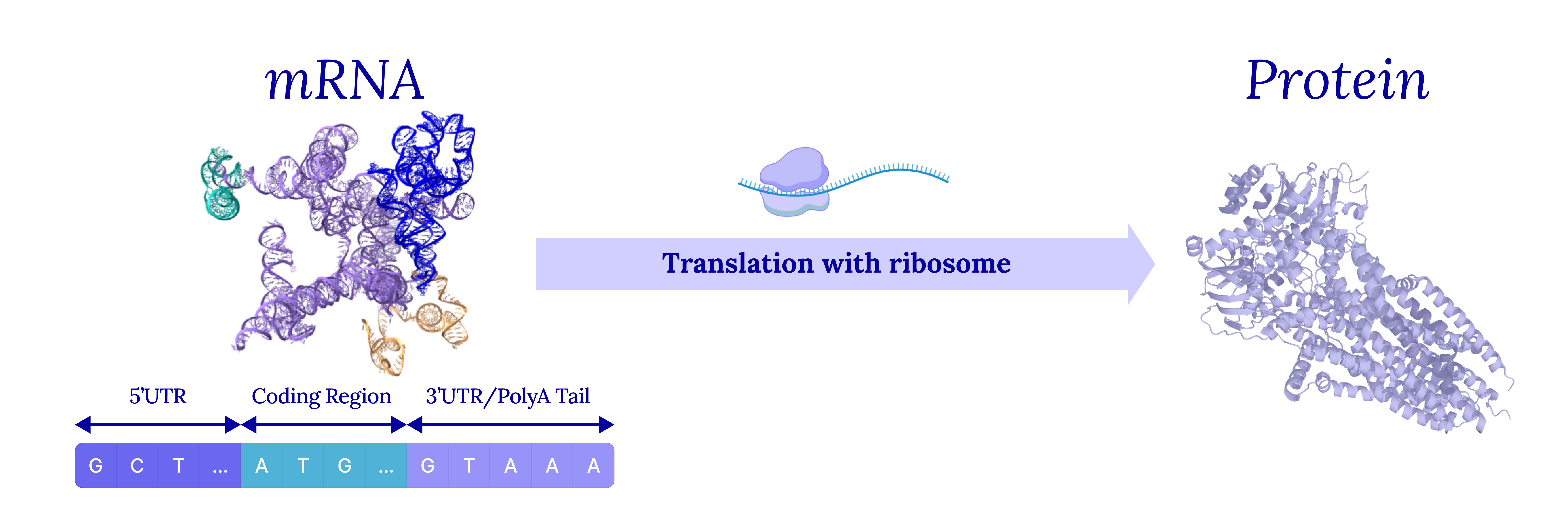
What makes mRNA so attractive as a therapeutic is exactly its position in the pipeline. If you can hand a cell a piece of mRNA encoding a protein of interest, you skip the genome entirely — no editing and no permanent change to the patient. The cell’s own translation machinery reads the mRNA, makes the protein for as long as the mRNA is intact (usually hours to days), and then degrades it. It is a tunable, transient way to dose any protein you want, to any organ reachable by the delivery system.
But a therapeutic mRNA isn’t just the coding sequence (CDS) for that protein. The full-length transcript has three functional regions concatenated together:
The 5’ untranslated region (5’UTR) recruits the ribosome and controls translation initiation.
The coding sequence (CDS) encodes the protein and is written in codons.
The 3’ untranslated region (3’UTR) controls mRNA stability, localization, and decay.
Each of these regions, along with their interactions, determines how stable the mRNA is, how efficiently it is translated, and how much protein is actually expressed in the patient. All three properties together determine the drug’s efficacy.
How mRNA is reshaping medicine
The notable advantage of mRNA is its versatility. Because it simply instructs the cell to make a protein, it applies to virtually any disease where a protein could be therapeutic.
Infectious disease vaccines. There are four FDA-approved mRNA vaccines on the market right now, and all of them happen to be respiratory virus vaccines. Three came out of the COVID-19 pandemic: BNT162b2 (Pfizer/BioNTech) and mRNA-1273 (Moderna), both encoding the full-length SARS-CoV-2 spike glycoprotein with two stabilizing proline mutations, and the newer mRNA-1283, which encodes only the receptor-binding and N-terminal domains of spike protein and can be dosed lower as a result. The fourth, mRNA-1345, prevents lower respiratory tract disease caused by respiratory syncytial virus (RSV).
Cancer vaccines. Cancer vaccine mRNAs encode neoantigens: short peptide fragments produced by mutations in the tumor’s own DNA, which appear on cancer cells but not on healthy cells and are typically unique to each patient’s tumor. Once the mRNA is translated, fragments of the neoantigen are displayed on MHC molecules at the cell surface, where they prime a T-cell response that attacks tumor cells carrying the same mutations. Several mRNA cancer vaccines are now in advanced clinical trials, with the most mature program targeting melanoma. Because every tumor has a unique set of mutations, mRNA’s rapid manufacturing timeline makes truly personalized vaccines feasible: sequence the tumor, identify the neoantigens, design and dose the mRNA. The same approach is now being tested for pancreatic cancer and glioblastoma.
Protein replacement therapies. For genetic diseases where a patient is missing a functional copy of a particular protein, mRNA can deliver the therapeutic protein in situ without permanently editing the genome. Compared to CRISPR-based therapies, mRNA replacement is less invasive, transient, and dramatically faster to manufacture. There are now mRNA candidates in clinical trials for pulmonary diseases, liver disease, and several rare metabolic conditions.
In vivo antibody production. Antibody manufacturing is slow and expensive. An emerging alternative is to deliver an mRNA encoding the antibody and let the patient’s own cells produce it. The first-in-human trial of an mRNA-encoded monoclonal antibody, Moderna’s mRNA-1944, targeting Chikungunya virus, demonstrated dose-dependent neutralizing antibody titers in healthy volunteers. A second clinical program, mRNA-6231, delivers an engineered IL-2 mutein to expand regulatory T cells for autoimmune indications.
The challenge: successful mRNA therapies are difficult to engineer
Loose mRNA in the bloodstream is rapidly degraded by nucleases, and the immune system is designed to recognize foreign RNA as a viral signature. To get any therapeutic mRNAs into a cell, you have to package them inside a lipid nanoparticle (LNP), which protects the RNA from degradation, masks it from the immune system, and delivers it across the cell membrane.
But even with LNP delivery, there remain several challenges to designing an mRNA that serves as a successful therapeutic. mRNAs are unstable, have transient expression profiles, and, depending on translation efficiency, often require high doses to achieve a clinical effect, which in turn drives up cost, manufacturing burden, and side-effect risk. Designing mRNA sequences with better stability or translation efficiency translates directly into lower doses, fewer side effects, and broader applicability. This is why we built mRNAutilus.
The Status Quo for mRNA Design
Before we get to what mRNAutilus does differently, let’s first establish the status quo of mRNA sequence design. Once a protein target is chosen (for example, SARS-CoV-2 spike protein), the sequence design problem breaks into three sub-problems:
Codon optimization: choosing which of the (typically 2–6) synonymous codons should encode each amino acid in the CDS.
UTR selection or design: picking 5’ and 3’ UTRs that maximize translation efficiency and stability.
Final assembly: concatenating the optimized CDS with the chosen UTRs into the final transcript.
The dominant approach to step 1 for the last ~40 years has been the Codon Adaptation Index (CAI), introduced by Sharp and Li in 1987. The CAI is essentially a lookup table: for each amino acid in your CDS, choose the synonymous codon that the host organism’s most highly expressed genes prefer. This works because cells maintain tRNA pools roughly proportional to codon usage, so “preferred” codons get translated faster.
CAI-based codon optimization is fast, simple, and the default in virtually every commercial mRNA design tool. However, it does not account for interactions between codons. The choice of codon at a single position can affect the local secondary structure, which in turn affects translation of the entire downstream sequence. Treating each codon in isolation against a static table ignores all of these interactions.
For UTRs, the default is even more standardized. The industry practice is to borrow UTRs from well-characterized natural genes, which are usually the 5’ and 3’ UTRs of human alpha-globin (HAB) that have evolved over hundreds of millions of years to be exceptionally stable and translatable in erythroid cells.
While HAB UTRs work surprisingly well across many contexts, UTRs co-evolve with their coding sequences, so pasting an alpha-globin 5’UTR in front of an arbitrary engineered CDS leaves a lot of room for potential optimization.
A wave of recent computational work has been introduced to improve this status quo. Predictive models (often pre-trained nucleic acid language models) can score candidate sequences for stability, translation efficiency, or expression. In silico screening pipelines generate thousands of candidate variants and rank them. Generative models have begun to design 5’UTRs alone, or 3’UTRs alone, or CDS alone, but almost always one component at a time, with the other components held fixed. The closest thing to a full-transcript generative method, GEMORNA, designs each component autoregressively in isolation, then concatenates them.
Gaps in Therapeutic mRNA Design
We can now make the three gaps in current mRNA design concrete, each of which mRNAutilus is built to address.
I. Lack of codon-UTR co-optimization
As discussed above, virtually every existing method designs the CDS and the UTRs separately, either by holding one fixed while optimizing the other, or by training separate models for each region. This wastes an important signal in the data: the interactions between regions.
Cross-region interactions between the 5’UTR, the CDS, and the 3’UTR jointly determine mRNA fitness. A 5’UTR that recruits the ribosome efficiently can be inhibited by a CDS that forms a secondary structure that blocks the start codon. A 3’UTR chosen for stability can have no effect when coupled with a coding sequence full of non-optimal codons, which slows the ribosome. Optimizing each region in isolation, then simply concatenating them together, fails to account for these interactions.
Our goal was to train a single generative model that could be handed a fully or partially masked transcript (any combination of masked UTRs and masked CDS positions) and fill in the missing nucleotides under a unified objective. UTR-only design, CDS-only design, and whole-transcript design should all be special cases of the same model, not three different pipelines.
II. Optimizing mRNA across multiple competing properties
mRNA therapeutic efficacy cannot be described with a single objective to optimize. It is multiple (often competing) properties at once. Half-life, translation efficiency, and protein abundance are not perfectly correlated, and the right tradeoffs depend on the application.
For instance, increasing translation efficiency can shorten half-life by speeding up the transcription rate. Stabilizing the 3’UTR for a longer half-life can block regulatory motifs that drive protein output. Focusing on only one of these objectives or collapsing them into a single objective ignores the trade-offs between objectives inherent in the design problem.
The right approach is Pareto optimization. Instead of returning the single “best” sequence (which doesn’t exist when objectives conflict), the model should return a set of non-dominated sequences, where no objective can be improved without sacrificing another. Drug developers can then pick sequences from the set based on the priority of each objective, given their application.3
III. Discrete generative modeling that captures long-range dependencies
Full-length mRNAs are long. A typical CDS is 1,000-3,000 codons, plus UTRs that can add another several hundred nucleotides on each side. Despite its length, interactions can occur at any pair of points along the sequence. Codon choice at one position can influence ribosome behavior thousands of bases downstream, and the 5’UTR motif can interact with a 3’UTR element on the opposite end of the transcript.
Autoregressive models, which currently dominate for language generation, are a poor fit in this setting. They generate one token at a time, left-to-right, which means decisions made early in the sequence cannot be revisited in light of later context. They also struggle to account for bidirectional dependencies over thousands of tokens.
Masked discrete diffusion models (MDMs)4 are a well-known solution to the limitations of autoregressive models. MDMs generate sequences by iteratively unmasking tokens from a sequence of absorbing [MASK] tokens, with an adaptive order of unmasking. At every step, the model sees the entire partially-unmasked sequence and predicts the token identity that the remaining masked positions should be, which gives it bidirectional context and the ability to reason about long-range dependencies. They have proven effective for protein and peptide design, and they are particularly well-suited for property-guided generation, because the iterative unmasking process is a natural place to inject external reward signals.
How mRNAutilus Addresses These Gaps
mRNAutilus has three key components: (1) a pretrained masked discrete diffusion model over full-length mRNAs, (2) a set of lightweight property regressors that score candidate sequences on therapeutic objectives, and (3) Monte Carlo Tree Guidance, which uses the regressors to steer the diffusion model toward Pareto-optimal sequences.
Trained on over 14 million mRNAs, with hybrid tokenization
We trained mRNAutilus on 14.2 million vertebrate mRNA sequences from the Ensembl genome browser, spanning 342 vertebrate species, which is one of the largest curated datasets of full-length mRNAs used for generative pretraining. After filtering for valid coding sequences, appropriate start/stop codons, and non-missing UTRs, we ended up with ~5.5 million sequences for the final training set.
The interesting design choice was tokenization. We use a hybrid scheme, where:
The CDS is tokenized by codon: 64 codons plus the start and stop tokens, treated as atomic units.
The 5’UTR and 3’UTR are tokenized by single nucleotide: A, C, G, T (plus standard IUPAC degenerate codes for ambiguity).
Tokenizing the CDS by codon means that synonymous-codon constraints can be enforced at sampling time to guarantee the encoded protein is preserved. Tokenizing the UTRs at single-nucleotide resolution preserves the model’s ability to design arbitrary regulatory sequences, including motifs that span codon-frame boundaries (which are meaningless in UTRs).
Architecturally, mRNAutilus is a 150M-parameter BERT-style transformer trained using the standard masked-diffusion cross-entropy objective.
In short, the model learns to take any partially-masked transcript and predict what the masked positions should be, given full bidirectional context over all tokens that are already unmasked. By varying the masking percentage during training, the model learns to complete anything from a 5%-masked sequence and a 90%-masked sequence, enabling accurate generation of mRNA within the underlying data distribution.
After pretraining, we checked that mRNAutilus had learned a faithful distribution of natural mRNAs by sampling sequences at lengths varying from 1,000, 2,500, to 5,000 tokens and comparing them against length-matched natural mRNAs. The generated sequences resemble natural statistics across all metrics: GC content converges to the natural distribution, Kozak consensus motif frequencies match, pairwise sequence diversity, and per-sequence Shannon entropy are essentially identical to natural sequences.
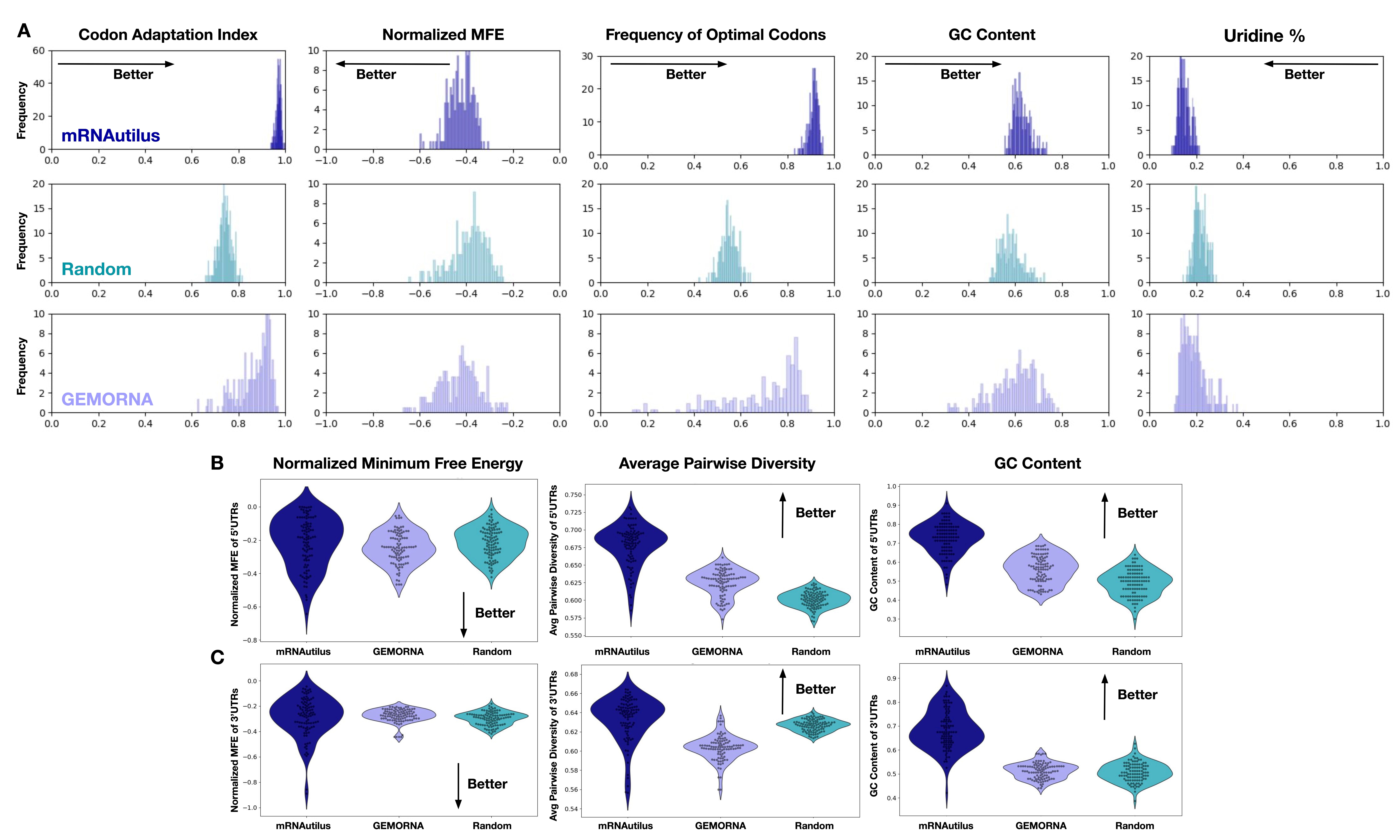
We show that mRNAutilus does not collapse into the repetitive-token failure modes that are common in generative language models. It actually generates more thermodynamically stable sequences than natural mRNAs, demonstrating that even unconditional generation yields useful structures.
Lightweight regressors as therapeutically relevant reward signals
Once we had a generative backbone, we needed a way to score candidate sequences. Since the model was trained by predicting the identities of masked tokens given the mRNA context, the pretrained model inherently learns rich representations of mRNA that should encode both coding structure and functional context.
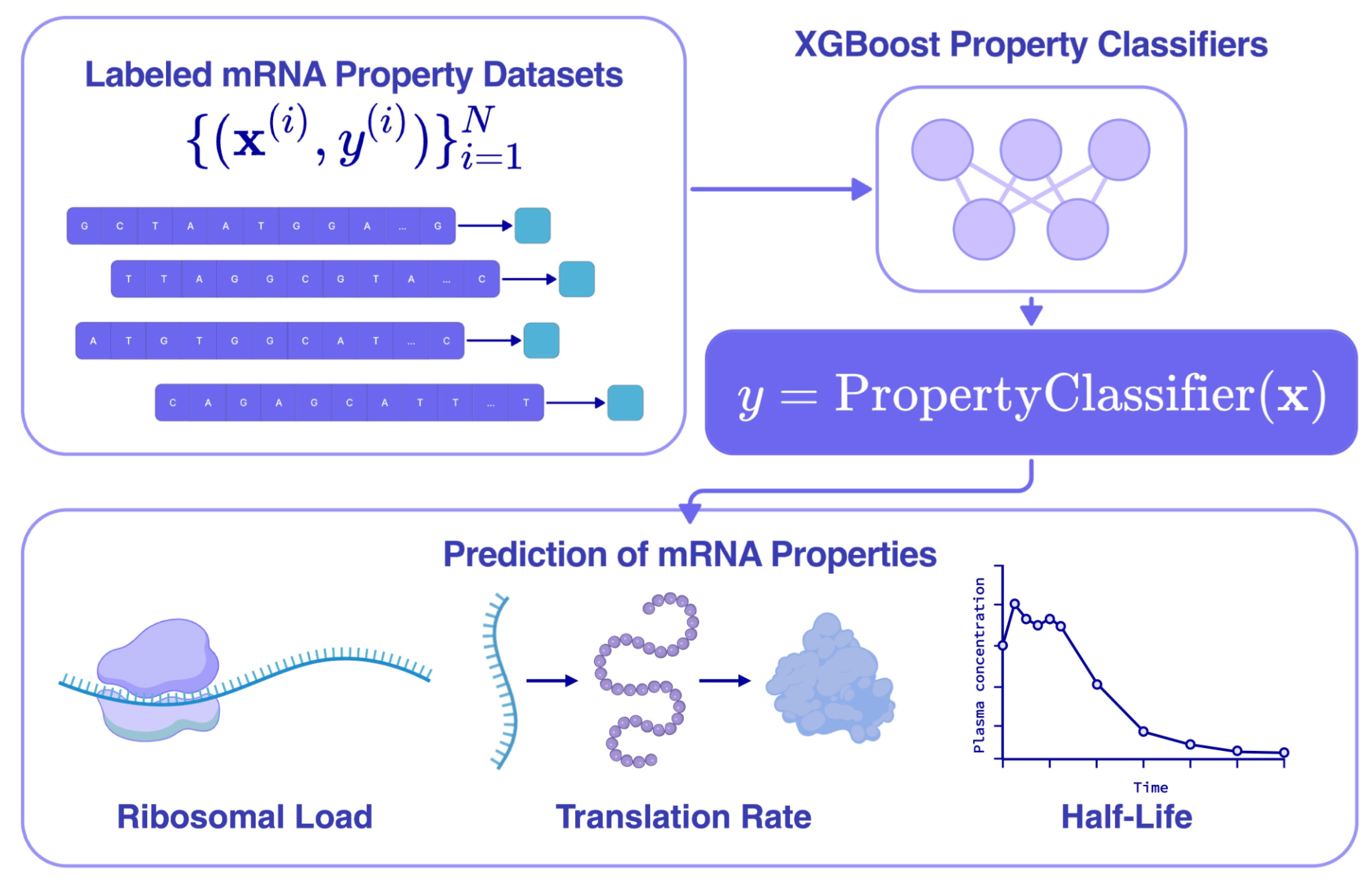
Leveraging these representations, we trained lightweight boosted tree regressors on the pooled per-token hidden embeddings to predict three properties central to therapeutic mRNA fitness from experimentally labeled data:
Half-life controls how long the transcript persists in the cell, and therefore the total protein output per dose.
Translation efficiency, measured by polysome profiling, controls how efficiently the ribosome translates each transcript.
Protein abundance, the ultimate functional readout, is the steady-state protein level produced by the mRNA.
All three regressors achieved strong validation R² on held-out data, and XGBoost consistently outperformed KNN-based alternatives on these tasks. When benchmarked against embeddings from other nucleic acid language models (Hyena-DNA, RiNALMo, Helix-mRNA, Evo-2), mRNAutilus embeddings achieved the highest validation R² on half-life prediction and were competitive on the other two, despite having ~45× fewer parameters than Evo-2 (150M vs. 7B).
The takeaway is that training a generative model with mRNA-specific tokenization can match or beat much larger general-purpose nucleic acid models on the specific tasks that matter for mRNA design.
These regressors, together with the pretrained backbone, give us everything we need to define the reward vector that drives multi-objective generation.
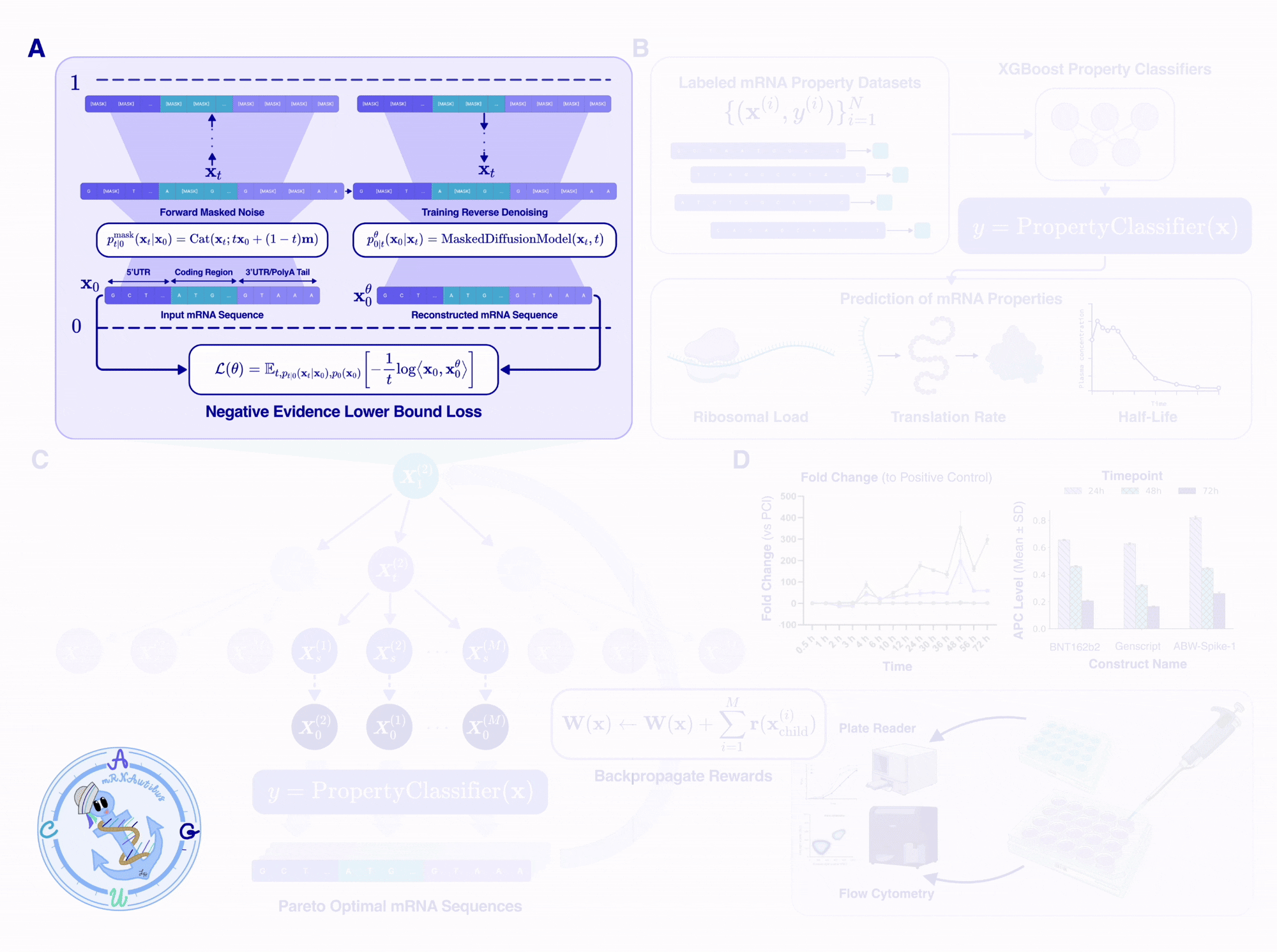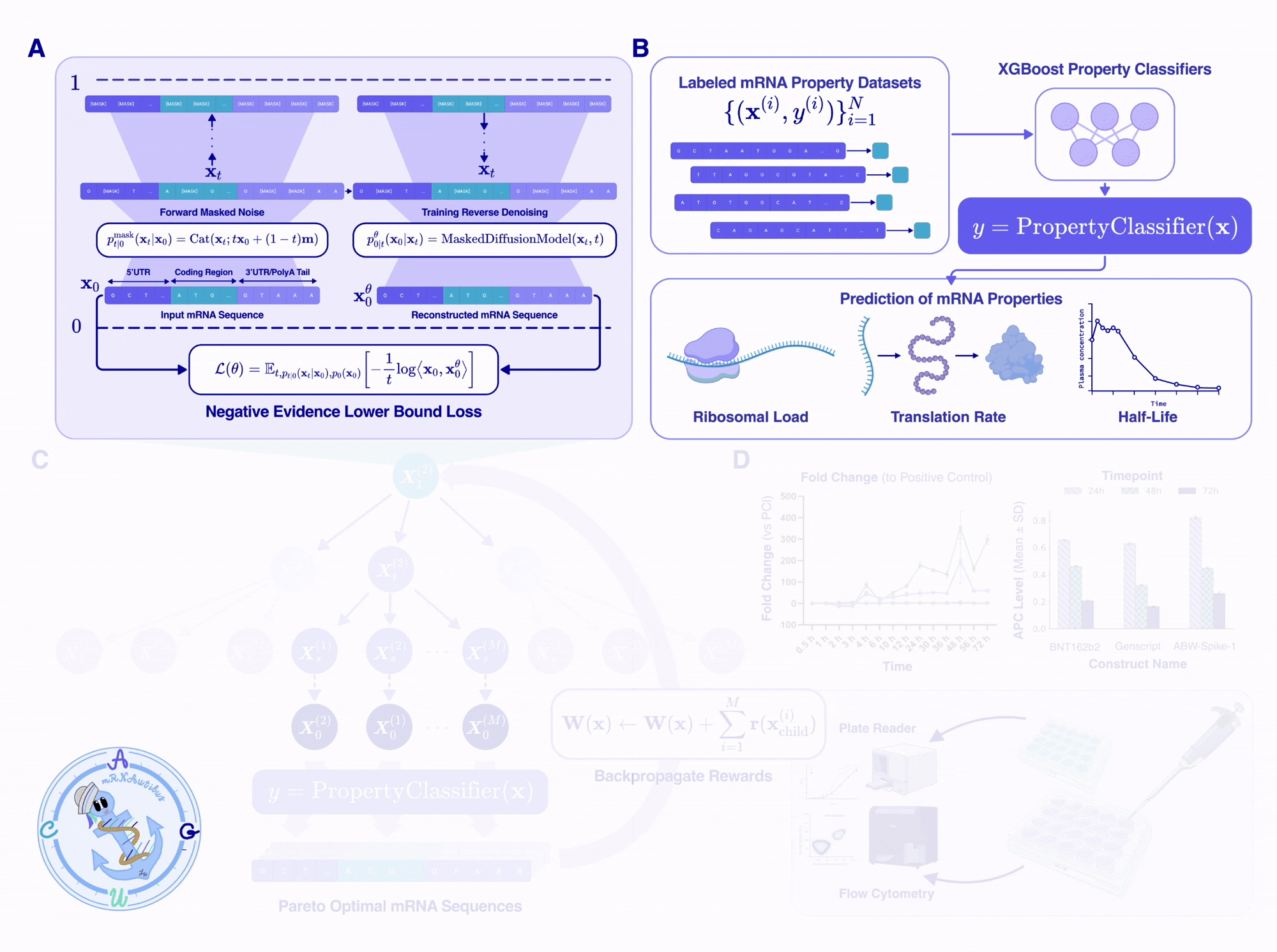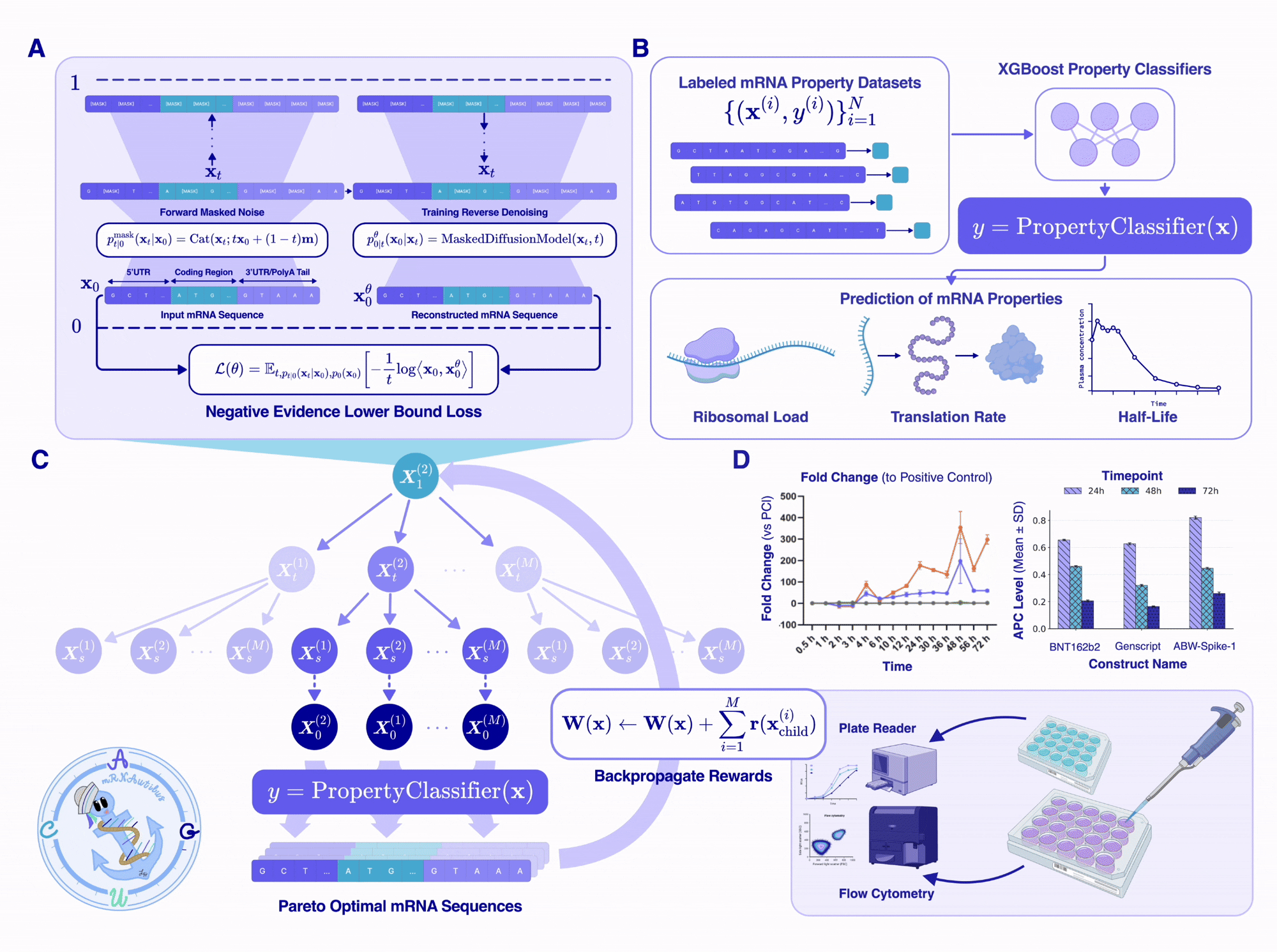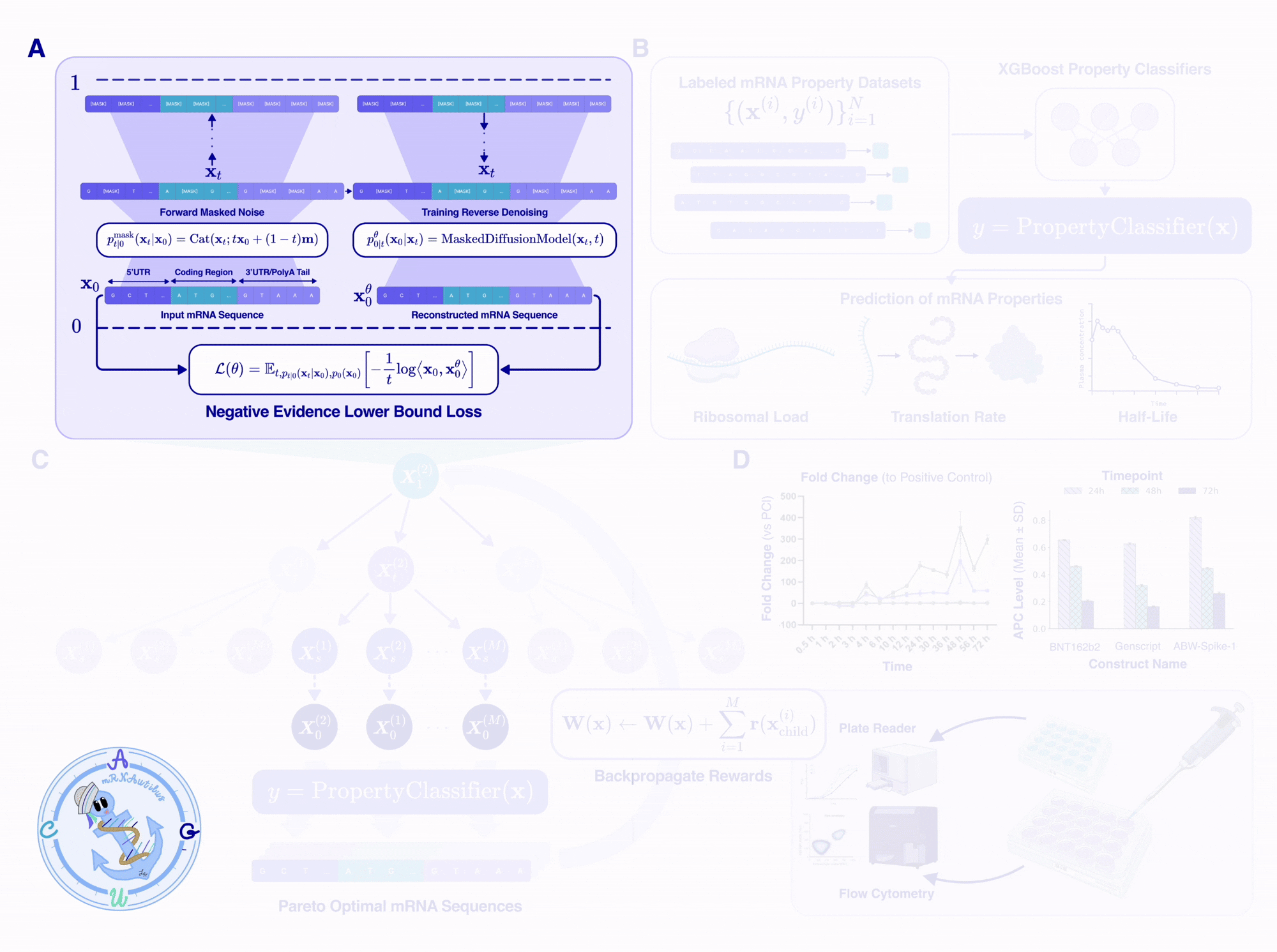
Monte Carlo Tree Guidance for multi-objective optimization
This is where the pieces come together. At generation time, we initialize a fully or partially masked mRNA transcript (masked UTRs and a masked or partial CDS) and use Monte Carlo Tree Guidance (MCTG)5 to iteratively unmask the sequence while maximizing multiple reward values.
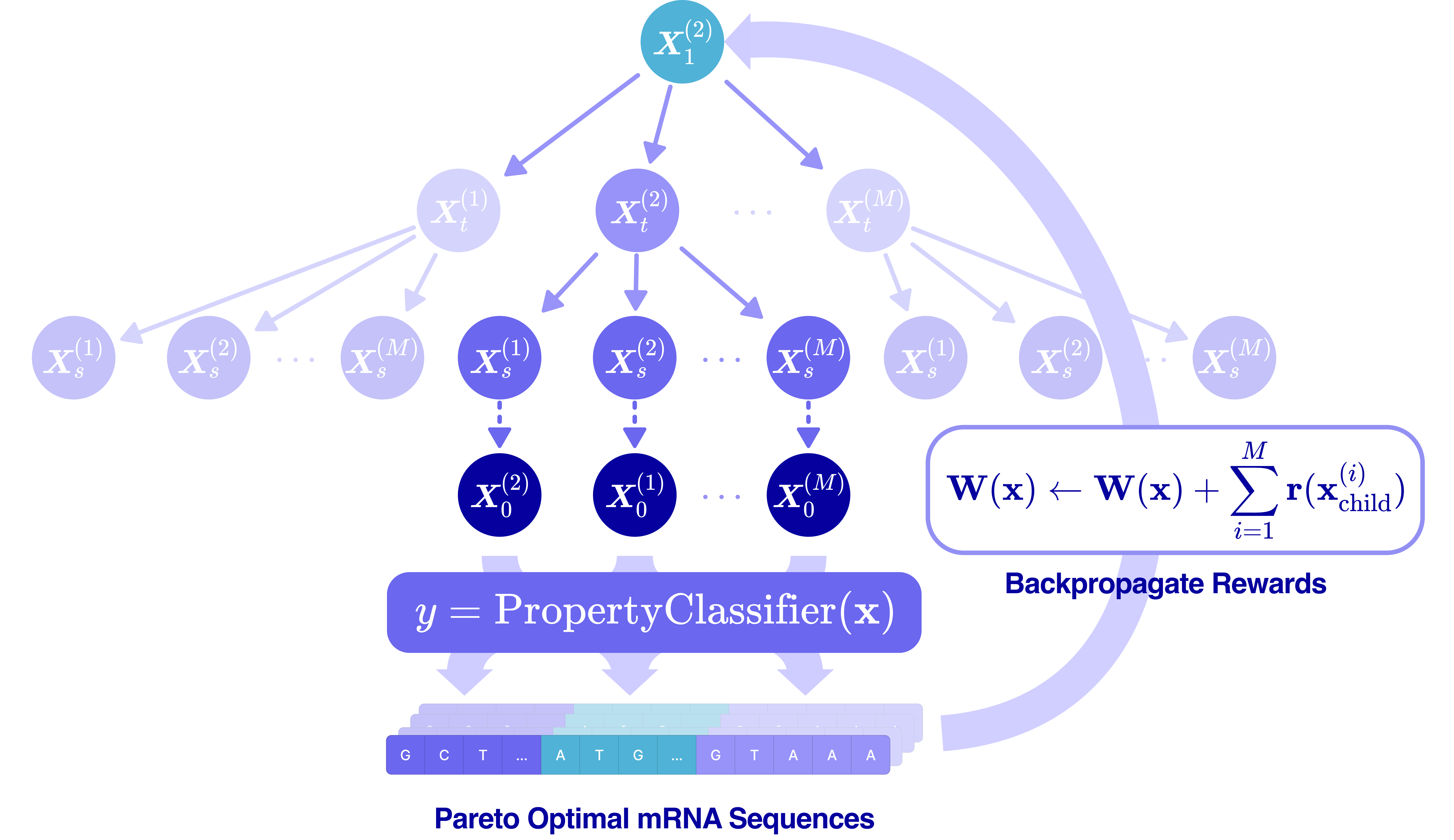
MCTG treats the multi-objective optimization as an iterative search procedure over a tree of unmasking trajectories, where each node is a partially-unmasked sequence, and each edge is an unmasking step. The algorithm builds and traverses the tree through four steps:
Selection. From the root (fully masked sequence), traverse the tree toward a leaf (partially masked sequence), choosing children that have either been visited infrequently (exploration) or have produced high-reward sequences in past rollouts (exploitation). Selection uses Pareto dominance over a per-objective score vector, so we are not collapsing rewards to a scalar at any point.
Expansion. At the chosen leaf, sample a set of distinct child sequences by adding Gumbel noise to mRNAutilus’s predicted denoising distribution and performing a set of unmasks corresponding to a single discrete time step for each child. Synonymous-codon constraints are enforced so that masked codon positions are only filled by codons that preserve the input template’s encoded amino acid, guaranteeing the protein sequence is conserved.
Rollout. For each child, complete the rest of the sequence using standard ancestral sampling for the remaining steps to get a fully unmasked transcript. Pass that transcript through the three property regressors to get a score vector. Add it to the global Pareto-optimal set if it is non-dominated6, and remove any sequences that it dominates.
Backpropagation. The reward for each child — which we define as the fraction of the current Pareto set that the child dominates, computed per objective — is propagated back up to all ancestor nodes in the tree, biasing future selections toward unmasking trajectories that yield Pareto-optimal sequences.
After a specified number of iterations, the algorithm returns the final Pareto-optimal set of sequences containing the mRNA designs that span the optimal trade-off space between half-life, translation efficiency, and protein abundance.
The advantage of MCTG is that it is gradient-free, modular, and operates entirely at inference time. To add a new objective, we do not need to retrain or fine-tune mRNAutilus, we can just add another regressor to the reward vector. We do not need labeled data conditioned on combinations of properties. We do not need smooth, differentiable rewards. We just need a black box function that scores a clean sequence on each objective separately, and the algorithm balances the combined trade-offs.
Using the MCTG algorithm, we designed full-length transcripts for firefly luciferase, SARS-CoV-2 spike protein, prime editing payloads, and engineered E3 ligases for proteome modulation, obtaining a Pareto frontier optimized mRNAs for experimental testing.
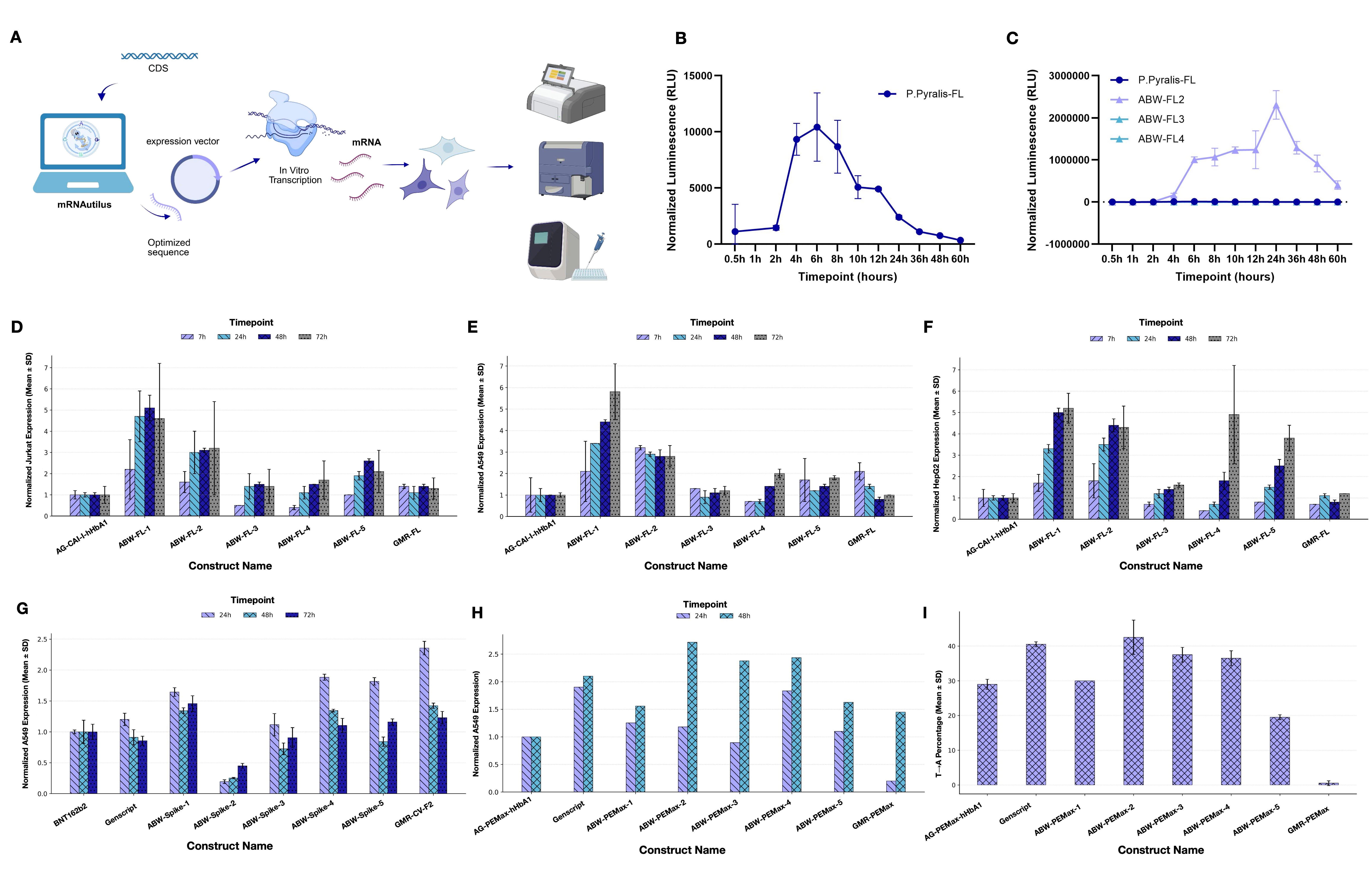
Testing mRNAutilus Designs in Cells
The stress test for any in silico design framework is putting the sequences into a cell and measuring protein output or downstream effects. We chose three targets that span the range of likely mRNA therapeutic applications:
Firefly luciferase (Fluc): the gold-standard reporter, useful because protein output is a luminescence readout we can quantify precisely across cell lines and time points.
SARS-CoV-2 spike glycoprotein: a real clinical antigen, where the benchmark is the actual BNT162b2 vaccine sequence and a commercial GenScript spike protein construct.
PEMax: a prime-editor payload, a genuinely demanding intracellular protein.
For each target, we ran MCTG with the half-life and translation efficiency regressors as the reward signal, generated 500 candidate sequences, and synthesized the top four by predicted fold-improvement over the human alpha-globin UTR baseline. None of these targets appeared in the mRNAutilus pretraining corpus or in the regressor training sets, making it a true test of de novo mRNA design with optimized therapeutic properties.
The results were remarkable. For luciferase, one zero-shot mRNAutilus design (ABW2) expressed at ~400-fold higher levels than wild-type Fluc at 48 hours post-transfection in HEK293T cells. The same designs outperformed a zero-shot GEMORNA-designed luciferase and a CDS+HAB-UTR composition across three additional cell lines (Jurkat, A549, HepG2).
For spike protein, three of the four mRNAutilus designs exceeded both BNT162b2 and the commercial GenScript construct in A549 cells, and matched the expression of GEMORNA’s lab-in-the-loop multi-shot design (GMR-CV-F2) — which had been iteratively refined experimentally — while one mRNAutilus design (ABW-Spike-1) showed improved intracellular stability over GMR-CV-F2.
The PEMax results were arguably the most demanding test. PEMax is an optimized prime editor that enables precise programmable genome edits without requiring double-strand breaks. Compared to a compact reporter like luciferase, it is a much harder design target as it consists of a long coding sequence, a complex domain architecture, and a functional readout (actual editing of an endogenous locus) that depends on the protein folding and localizing correctly inside the cell.
We demonstrate that several mRNAutilus-designed PEMax constructs matched or exceeded the commercial GenScript PEMax mRNA in expression. The best mRNAutilus design, ABW-PEMax-2, achieved the highest T to A editing efficiency of any construct tested, with two other mRNAutilus designs editing at comparable rates. By contrast, PEMax sequences designed with GEMORNA showed barely any editing. Across these diverse therapeutic payloads, the pattern is clear: zero-shot mRNAutilus generation produces mRNAs with superior expression and durability to baseline methods.
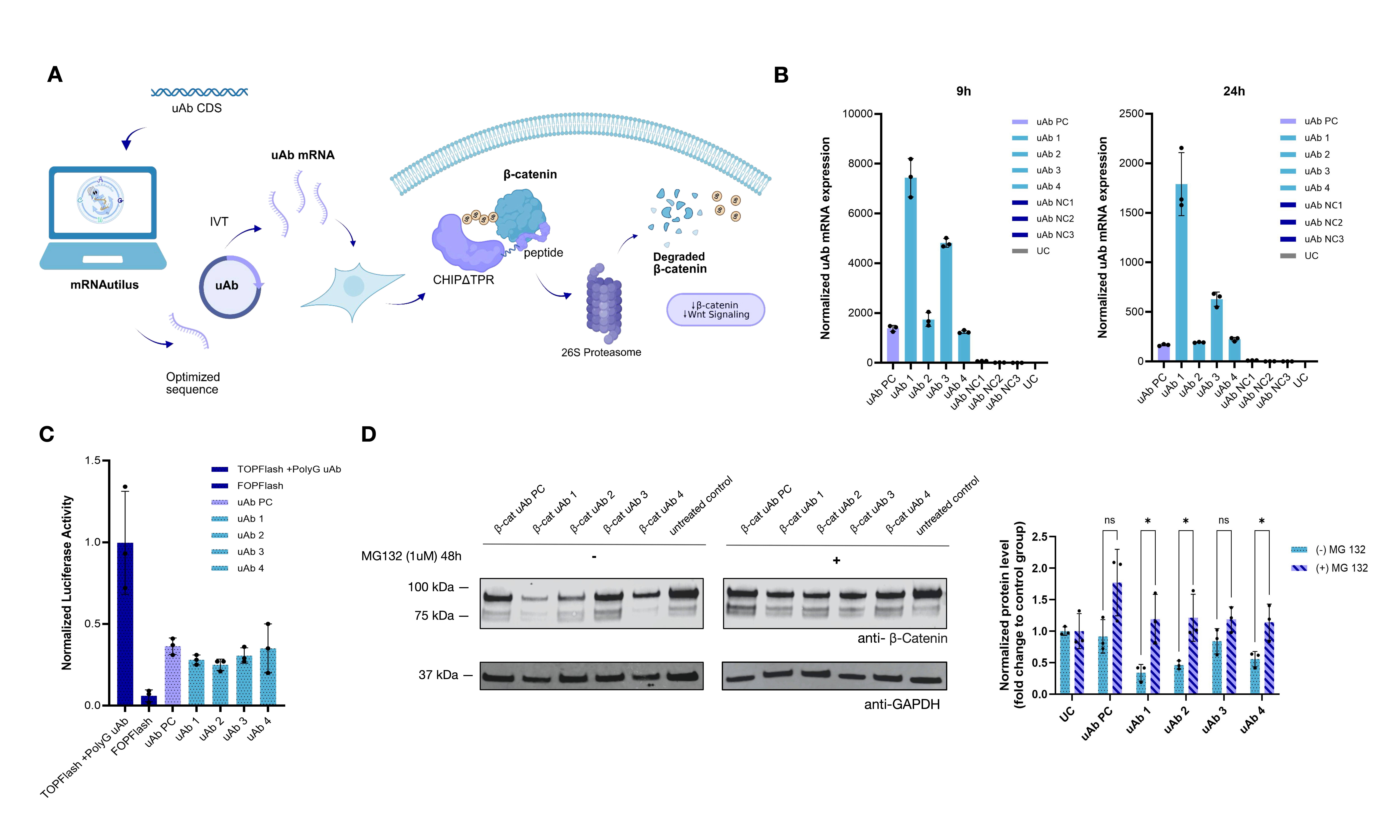
The most interesting test, though, was a payload that has yet to be approved as a drug: a ubiquibody (uAb) targeting β-catenin.
uAbs and programmable protein degradation
Ubiquibodies (uAbs) are engineered E3 ubiquitin ligases. In a uAb, the natural substrate-binding domain of CHIP (a host E3 ligase) is replaced with a language-model-designed peptide binder, redirecting the cell’s own ubiquitin-proteasome machinery to degrade a target of choice. They are particularly attractive as mRNA payloads because mRNA-LNP delivery enables transient, tunable intracellular expression of the degrader without genomic integration.
We targeted β-catenin — a driver of Wnt-pathway oncogenesis whose stabilization promotes c-Myc and cyclin D-1 expression — using a previously validated peptide binder from PepPrCLIP. We then used mRNAutilus to design the mRNA encoding the uAb.
The results, in DLD-1 colorectal cancer cells, were significant:
Optimized uAb mRNAs showed markedly higher transcript abundance at 9h and 24h post-transfection than the human alpha-globin baseline (qPCR readout).
TOPFlash Wnt/β-catenin reporter assays showed that the optimized uAbs fully retained their intended activity, suppressing Wnt signaling — some designs more strongly than the positive control.
Immunoblotting of cytosolic fractions confirmed significant reductions in endogenous β-catenin, and crucially, these reductions were abolished by the proteasome inhibitor MG132 — confirming that the observed degradation goes through the ubiquitin-proteasome pathway, not some off-target effect.
The last point demonstrates that mRNAutilus optimization not only produced more transcripts, but it also produced more functional intracellular degraders on an endogenous oncogenic target.
These results demonstrate that mRNAutilus is broadly applicable, not just to reporters and antigens, but to programmable proteome modulation as a therapeutic modality.
Closing Thoughts
mRNAutilus is a significant step toward a unified framework for controllable, multi-objective-guided, full-length mRNA therapeutic design, and it opens the door for several follow-up directions that I wanted to highlight here.
On the data side, property guidance is only as good as the trained predictors, and public mRNA datasets remain sparse, cell-type-restricted, and heterogeneous in their experimental readouts. The most impactful thing the field could do right now is generate large, uniformly measured datasets across half-life, translation efficiency, protein abundance, immunogenicity, and tissue-specific expression.
On the application side, mRNAutilus can readily be expanded beyond reporters, antigens, prime editors, and engineered E3 ligases to mRNAs encoding anything from enzymes, antibodies, cytokines, and the next generation of programmable proteome modulators. Paired with LNP delivery, mRNAutilus-designed transcripts should enable transient, tunable, and target-specific modulation of intracellular proteomes for a broad set of currently undruggable diseases.
mRNAutilus holds a special place in my heart because it builds on one of my very first algorithms, MCTG from PepTune, and shows that it works in vitro for real, therapeutically relevant tasks.
This project would not be possible without my incredible co-authors: Sawan Patel, who led the computational development and organized both sides of the project, Yesol Kim, who led the experimental validation, Yinuo Zhang and Divya Srijay, our advisors Sherwood Yao and Pranam Chatterjee, and our collaborators from GenScript!
Read the full paper on arXiv, or try it yourself on the mRNAutilus interface at autona.atombio.ai/home.
Citation
If you find this article helpful for your publications, please consider citing our paper:
@article{patel2026mrnautilus,
author = {Patel, Sawan and Tang, Sophia and Kim, Yesol and Zhang, Yinuo and Srijay, Divya and Lin, Ping-Jung and Shubham, Shambhavi and Pi, Fengmei and Wu, Cedric and Yao, Sherwood and Chatterjee, Pranam},
title = {mRNAutilus: Multi-Objective-Guided Discrete Generation of mRNA with Optimized Therapeutic Properties},
journal = {arXiv preprint arXiv:2605.31296},
year = {2026},
}Alchemy Bio is a blog where I share transformative ideas in machine learning for biology. If you don’t want to miss upcoming posts, you should consider subscribing for free to have them delivered to your inbox.
Some of my early research has actually been in designing targeted LNP platforms that deliver mRNA therapeutics to the brain!
For context: mRNAutilus (pronounced M-R-Nautilus) stands for “mRNA generation via unrolled trajectories and informed latent updates.” And it sounds like Nautilus.
This is the same idea we used in PepTune for multi-objective therapeutic peptide design, and it adapts naturally to mRNA.
…which I described in more detail in my previous article.
…meaning that no other sequence in the set has better or equivalent values across all rewards and a strictly better value on at least one reward.
 | A guest post by
|